|
 |
|
|
|
|
|
|
|
La teoria mitocondriale dell’invecchiamento ritiene che come noi viviamo e produciamo ATP i nostri mitocondri producono radicali liberi che inesorabilmente attaccano i nostri mitocondri e producono mutazioni nel nostro DNA mitocondriale somatico. In persone la cui produzione energetica era già compromessa (sia con mitocondri ereditati mutati o mutazioni nucleari o per tossine o per altri fattori) il danno risultante dal DNA mitocondriale porterà molto più facilmente alla produzione di un livello di energia inferiore al desiderabile. Questi individui mostreranno sintomi precoci che progrediranno fino ad un completo quadro patologico molto più rapidamente di quelle persone che inizialmente non avevano deficit nella loro capacità di produzione energetica.
D I S O R D I N I M I T O C O N D R I A L I Encefalomiopatia mitocondriale
Malattia Di Alpers - Polidistrofia
cerebrale progressiva infantile. Polidistrofia infantile progressiva. Sintomi: attacchi, demenza, spasticità, cecità, disfunzioni epatiche e degenerazione cerebrale. La malattia di Alpers può essere causata da disturbi della fosforillazione ossidativa, includendo la sindrome di DNA mitocondriale impoverito
Sindrome Di Barth - Cardiomiopatia Infantile
Mortale (una cardiopatia di cui sono stati documentati solo 15 casi in tutto il mondo di cui due nel nostro paese) La sindrome di Barth è una malattia metabolica caratterizzata da cardiomiopatia dilatativa, più raramente di tipo ipertrofico, neutropenia, miopatia scheletrica, difetto di crescita. Tuttavia il quadro clinico può avere un'espressione variabile e si può presentare progressivamente o all'improvviso. Nella maggior parte dei casi la malattia si manifesta durante l'infanzia. La presentazione iniziale più comune è l'arresto cardiaco. La sindrome di Barth sembra molto rara, ma può colpire tutti i gruppi etnici. Successivamente alla sua caratterizzazione clinica e biochimica, il gene-malattia è stato mappato in Xq28, utilizzando il clonaggio posizionale; l'analisi mutazionale ha identificato il gene G4,5. Sono state identificate oltre 20 mutazioni. Il trattamento è essenzialmente di supporto e la presa in carico dovrebbe essere multidisciplinare, in particolare dovrebbe essere svolta sia dai cardiologi che dagli ematologi.
Mancanza Della Carnitina: Malattie con disturbo della demolizione degli acidi grassi - Codice di esenzione: RCG070 - Carenza della CPT (carnitin-palmitin-trasferasi) Difetto enzimatico che porta a diverse malattie 1) Malattia infantile, che può portare a carenza di zuccheri e ad arresto cardiaco improvviso. 2) Malattia dell’età adulta, che appare come debolezza muscolare e la colorazione marrone delle urine dopo sforzo fisico o digiuno; ereditabile sia dal padre che dalla madre (autosomale recessiva). L’andamento delle malattie mitocondriali è molto diversificato: Alcuni pazienti hanno un peggioramento occasionale (p.es. carenza di CPT). In alcuni pazienti i sintomi peggiorano molto lentamente (p.es. OECP più). La malattia può progredire anche molto rapidamente, specialmente se i sintomi subentrano nell’età infantile.
Mancanza Del Complesso I
- Sintomi: Tre forme maggiori: 1. Disordine multisistemica infantile fatale, caratteritardo da ritardo nello sviluppo, debolezza muscolare, malattia cardiaca, acidosi lattica congenita, e insufficienza respiratoria. 2. Miopatia che inizia nell'infanzia o nell'età adulta manifastantesi come debolezza o intolleranza all'esercizio. comune acido lattico elevato. 3. Encefalomiopatia mitocondriale (includendo MELAS), che può iniziare nell'infanzia o nell'età adulta e consiste di una variabile combinazione di segnali e sintomi, includendo oftalmoplegia, attacchi, demenza, atassia, perdita dell'udito, retinopatia pigmentosa, neuropatia sensoria, e movimenti incontrollabili. può causare la sindrome di Leigh. Trattamento: Somministrazione di tiamina, riboflavina, biotina, carnitina, co-enzima Q10, e dieta chetogenica hanno avuto successo in alcuni casi, ma non nella sindrome infantile multisistemica. Mancanza Del Complesso II - Carenza di succinato deidrogenasi. Sintomi: Encefalomiopatia e varie manifestationi, includendo la diminuizione della crescita, sviluppo ritardato, ipotonia, letargo, insufficienza respiratoria, atassia, mioclone. Comune l'acidosi lattica. Può causare la sindrome di Leigh. Mancanza Del Complesso III - Carenza di Ubiquinone-citocromo c ossidoriduttasi. Sintomi: Quattro forme maggiori: 1. Encefalomiopatia infantile fatale, acidosi lattica congenita, ipotonia, postura distrofica, attacchi, e coma. Comune ragged red fiber. 2. Encefalomiopatia ad insorgenza tardiva (dall'infanzia all'età adulta): varie combinazioni di debolezza, bassa statura, atassia, demenza, perdita dell'udito, neuropatia sensoria, retinopatia pigmentosa, e segni piramidali. Comune ragged red fiber. Possibile acidosi lattica. 3. Miopatia, con intolleranza all'esercizio che evolve in debolezza fissa. Comune ragged red fiber. Possibile acidosi latica. 4. Cardiomiopatia infantile istiocitoide. Mancanza Del Complesso IV/Mancanza Del Cox - Carenza di citochromo c ossidasi è causata da un difetto nel complesso IV della catena respiratoria. Sintomi: Due forme maggiori: 1. Encefalomiopatia: tipicamente normale per i primi da 6 a 12 mesi di vita poi appaiono regressione dello sviluppo, atassia, acidosi lattica, atrofia ottica, oftalmoplegia, nistagmo, distonia, segni piramidali, e problemi respiratori. Frequenti attacchi. Può causare la sindrome di Leigh. 2. Miopatia: Due varianti principali: a) Miopatia infantile fatale: può iniziare subito dopo la nascita ed accompagnarsi con ipotonia, debolezza, acidosi lattica, ragged red fiber, insufficienza respiratoria, e problemi renali. b) Miopatia infantile benigna: può iniziare subito dopo la nascita ed accompagnarsi con ipotonia, debolezza, acidosi lattica, ragged red fiber, problemi respiratori, ma (se il bambino sopravvive) seguirà un miglioramento spontaneo. Mancanza Complessa Di V - Mancanza del Trifosfato di adenosina Synthase:Sindrome della oftalmoplegia esterna progressiva cronica. La combinazione di ptosi e limitazione dei movimenti degli occhi è riferita all'oftalmoplegia. Sintomi: Miopatia lenta progressiva CPEO - Sindrome Esterna Progressiva Cronica Di Oftalmoplegia: Eredità mendeliana - materna - sporadica . Codice di esenzione: RFG110 Sindrome oftalmoplegica esterna cronica progressiva. Sintomi: Simili a quelli del KSS, più: miopatia visuale, retinite pigmentosa, disfunzioni del sistema nervoso centrale. Causa: Lacuna singola del DNA mitocondriale. Mutazione puntiforme del DNA mitocondriale: A3243G (la più comune) L'oftalmoplegia cronica è caratterizzata da una debolezza progressiva dei muscoli oculari e del muscolo elevatore della palpebra superiore e colpisce principalmente gli adulti. Può essere totalmente e permanentemente isolata. Tuttavia, in una bassa percentuale di casi, può associarsi una miopatia scheletrica, che coinvolge per lo più i muscoli assiali e prossimali, provocando un affaticamento anomalo e una debolezza muscolare permanente. In questo caso la malattia viene ancora definita oftalmoplegia esterna progressiva isolata. Numerose oftalmoplegie sono associate ad altri sintomi, confermando il pattern multisistemico di questa malattia. I sintomi associati sono eterogenei: neurologici (perdita dell'udito, retinopatia, difetti cerebellari, neuropatia periferica,ecc.), endocrini ( diabete, ipogonadismo, ipoparatiroidismo, ecc.), renali (insufficienza renale, tubulopatia, ecc.), cardiaci (difetti di conduzione, miocardiopatia, ecc.). L'associazione di vari sintomi porta all'identificazione di sindromi associate ad oftalmoplegia, come la sindrome di Kearns Sayre o sindrome MNGIE. Le cause dell'oftalmoplegia cronica sono diverse. La maggior parte di questi quadri clinici è dovuta a mitocondriopatie, ma la causa della disfunzione mitocondriale è variabile (mutazioni puntiformi, delezioni del DNA mitocondriale, mutazione di geni nucleari, con effetti sul DNA mitocondriale, come la timidina fosforilasi nella MNGIE). Altre cause sono la miastenia classica, la distrofia oculo-faringea e le sindromi miasteniche congenite. Tutte e tre le patologie determinano sintomi muscolari isolati, in assenza di difetti multisistemici. Tuttavia le malattie mitocondriali causano sia forme strettamente muscolari, sia forme generalizzate e multisistemiche MELAS - Codice di esenzione: RN0710 - Nella sindrome di MELAS (Encefalomiopatia mitocondriale con Acidosi Lattica ed episodi di ictus ), i sintomi clinici insorgono durante l'infanzia o nell'adulto prima dei 40 anni e possono essere scatenati da un'infezione o da uno sforzo fisico. Ai sintomi si associano, in modo più o meno incostante, emiparesi, emianopsia, deficit motorio, emicrania, vomito, demenza, convulsioni, sordità, diabete, disturbi della memoria. Spesso la famiglia dimostra un'eredità materna. Dalla biopsia muscolare si notano numerose ragged red fibers (fibre rosse ``stracciate''), fibre positive al citocromo ossidasi, un deficit dei complessi I e IV della catena respiratoria. Numerosi segnali iperintensi sono presenti nella sostanza bianca e del cervello. Nel sangue si trovano livelli elevati di lattato. La mutazione più frequente è A3243G del tRNA per la Leucina del DNA mitocondriale. * autrice: Dott.ssa C. Marsac (settembre 2002 KSS - sindrome di Kearns-Sayre (spodadica) - Codice di esenzione: RF0020 - Insorge prima dei 20 anni. Questo disordine è definito da PEO (solitamente come il sintomo iniziale). Comporta oftalmoplegia, ptosi, retinite pigmentosa, segni associati a miopatia, disturbi della conduzione cardiaca, iperproteinorrachia. Nei casi sporadici possono essere presenti altri sintomi come sordità, cardiomiopatia, crisi epilettiche, disturbi del transito intestinale, ipoparatiroidismo, ritardo nella crescita, diabete, insufficienza renale. Queste patologie sono associate ad ampie delezioni eteroplasmiche del DNA mitocondriale, la più frequente delle quali è lunga 4,9 kilobasi. Il rapporto tra la percentuale degli organelli mutati/non mutati può essere molto elevato e quando raggiunge la soglia del 60% circa (ad es: nel muscolo) determina un fenotipo patologio con deficit a carico dei complessi della catena respiratoria, anomalie istochimiche, "Ragged Red Fibers''. Alcune rare sindromi di Pearson possono evolvere in una sindrome di Kearns-Sayre.
NARP - Inizio: Infanzia - eredità materna Codice di esenzione: RF0030 La sindrome Neuropatia, Atassia e Retinite Pigmentosa (NARP) è clinicamente eterogenea, ma spesso è caratterizzata dalla combinazione tra neuropatia sensoriale-motoria, atassia cerebellare e cecità notturna. La sua prevalenza è stimata in circa 1/12.000. La NARP di solito colpisce i giovani adulti. La sintomatologia clinica comprende: retinopatia precoce "sale e pepe''; retinite pigmentosa; pupille "pigre''; nistagmo; cecità; debolezza muscolare prossimale; ritardo di sviluppo; atrofia cortico-spinale; demenza; ipoacusia; convulsioni; atassia; neuropatia sensoriale; debolezza muscolare prossimale neurogena. La sindrome NARP è una malattia ad eredità materna, dovuta alla mutazione 8993T>G nel gene del mtDNA, MTATP6, che codifica per la subunità 6 dell'ATPasi. La mutazione 8993T>G comporta un cambio aminoacidico dalla leucina 156, altamente conservata, in arginina (L156R), e causa un grave deterioramento della sintesi dell'ATP mitocondriale, che riduce l'energia e produce la morte cellulare, in particolare nei tessuti che dipendono fortemente dal metabolismo della fosforilazione ossidativa, come il cervello e la retina. La mutazione è anche presente nell'8-10% dei pazienti con malattia di Leigh, che, in questo caso, viene per convenzione definita con l'acronimo MILS (sindrome di Leigh ad eredità materna = maternally-inherited Leigh syndrome). La MILS è pertanto la forma clinica più grave della sindrome NARP e, di solito, si manifesta in generazioni successive affette, che simulano una pseudo-anticipazione. Il trattamento è solo sintomatico.
Sono stati recentemente proposti farmaci antiossidanti in base ad
evidenze sperimentali in vitro. MILS - Sindrome di Leigh ereditata per via matrilineare Codice di esenzione: RF0030
MERFF - Epilessia mioclonica, con le
fibre ragged-rosse - Eredità materna o sporadica -
Codice di esenzione: RF0020 -
Causa: Mutazioni mitocondriali del punto del DNA:
A8344G, T8356C Sindrome di Pearson: Eredità sporadica - Inizio: Infanzia - Codice di esenzione: RN1600 - è una rara malattia sporadica della primissima infanzia caratterizzata da anemia sideroblastica, pancitopenia ed insufficienza del pancreas esocrino con malassorbimento intestinale. Nei bambini che sopravvivono oltre i primi anni si assiste ad un progressivo miglioramento della situazione ematologica e gastrointestinale, mentre insorgono i segni caratteristici della sindrome di Kearns-Sayre (KSS) MDS La sindrome da deplezione del DNA mitocondriale - Codice di esenzione: RN0710 -
La sindrome MDS comprende un gruppo clinicamente eterogeneo di malattie mitocondriali, caratterizzate da una riduzione del numero delle coppie del DNA mitocondriale nei tessuti affetti, senza mutazioni o riarrangiamenti nel mtDNA. Per questo la MDS si considera un processo primitivo. La MDS può essere una patologia neurogena relativamente frequente nel periodo neonatale e nell'infanzia. In una casistica, l'11% dei bambini (<2 anni), che presentavano debolezza, ipotonia e ritardo dello sviluppo, avevano una deplezione di mtDNA. La MDS è eterogenea a livello fenotipico e si può manifestare in forma epato-cerebrale, miopatica, miopatica benigna ad esordio tardivo o cardiomiopatica. I bambini di solito presentano nell'infanzia ipotonia, acidosi lattica e livelli elevati di creatin-chinasi sierica. Alcuni di loro hanno anche una epatopatia grave, spesso fatale, oppure un coinvolgimento renale, che simula la sindrome di De Toni-Fanconi. La diagnosi clinica viene confermata dalla ridotta attività della catena respiratoria e, soprattutto, da un basso rapporto mtDNA/nDNA (DNA nucleare) nei tessuti affetti. La MDS spesso ricorre nelle famiglie, con una possibile trasmissione autosomica recessiva, a sostegno di un difetto genetico del nDNA. In effetti, le mutazioni nei geni della chinasi della deossiguanosina mitocondriale, codificata nel nucleo, e della chinasi della deossitimidina sono state associate rispettivamente alle forme epato-cerebrali e miopatiche della MDS. Questi due geni codificano enzimi per la via di salvataggio mitocondriale, implicata nella sintesi del mtDNA attraverso il rifornimento di deossiribonucelotidi. La debolezza muscolare e l'intolleranza all'esercizio possono rispondere all'integrazione con coenzima Q
MNGIE : - Codice di esenzione: RN0710 - Encefalopatia Mitocondriale Neuro Gastrointestinale . Questo disordine causa PEO, ptosis, debolezza degli arti e problemi (digestivi) gastrointestinali, compreso diarrea cronica e dolore addominale. Un altro sintomo comune è la neuropatia periferica Eredità mendeliana recessina Compare in genere tra i 10 e i 40 anni ed è caratterizzata dall'associazione di: oftalmoplegia cronica progressiva, neuropatia periferica, alterazioni della sostanza bianca cerebrale e disturbi gastrointestinali (episodi ricorrenti di diarrea e/o vomito causati da una motilità intestinale anormale). I pazienti in genere sono molto magri e bassi. Lo studio morfologico dei loro muscoli ha rilevato una piccola porzione di fibre muscolari "ragged-red'', dimostrazione di una proliferazione di mitocondri. L'analisi biochimica della catena respiratoria dei pazienti dimostra deficienze parziali in uno o più complessi respiratori. L'analisi mediante Southern blotting del DNA del muscolo documenta diverse forme di DNA mitocondriale deleto, che rappresentano tuttavia solo una piccola frazione del DNA mitocondriale totale. Una parziale mancanza di DNA mitocondriale può coesistere con delezioni multiple. Possono essere presenti altri sintomi, ma il loro riscontro non è necessario per la diagnosi della malattia. Fra essi, la perdita dell'udito, la retinite pigmentosa, difetti cerebellari, ecc. La MNGIE è trasmessa come carattere autosomico recessivo; il gene responsabile è localizzato sul cromosoma 22q13.32 e codifica per una timidina fosforilasi. La sindrome può essere identificata sia dosando l'attività della timidina fosforilasi nei leucociti, sia mediante l'analisi del gene stesso. Il nesso fisiopatologico tra questo difetto enzimatico e i sintomi della sindrome è ancora sconosciuto. Le terapie che riducono la timidina plasmatica possono dare beneficio, tuttavia non è stato ancora messo a punto un trattamento efficace. Mancanza di CPT I Sintomi: Fegato dilatato e ricorrenti episodi Reye-simili provocati da digiuno o malattie. Causa: Autosomale recessiva. Trattamento: Trigliceridi a media catena. Mancanza di CPT II Sintomi - Miopatica: Intolleranza all'esercizio, intolleranza al digiuno, dolore muscolare, rigidezza muscolare, e mioglobina nelle urine. Sintomi - Infantile: sindrome simil-Reye, fegato dilatato, ipoglicemia, cuore ingrandito, e aritmia cardiaca. Causa: Autosomale recessiva. Trattamento - Non-Infantile: dieta con molti carboidrati e pochi grassi. Acidosi lattica : l'acidosi lattica implica un metabolismo danneggiato, ma non è, in se stessa, diagnostico per una malattia mitocondriale. La acidosi lattica è un dato di quasi costante riscontro nel bambino "mitocondriale", mentre nell'adulto può essere assente. Acido in cui, nel tessuto muscolare, si trasforma l'acido piruvico, quando quest'ultimo non può entrare nei mitocondri ed essere ossidato, consentendo un corretto processo di respirazione delle cellule. Valori abnormi di acido lattico indicano un'alterazione dell'attività mitocondriale. La spettroscopia per risonanza magnetica fosforo-nucleare (P-NMR) è un altro metodo per testare la miopatia mitocondriale in vivo. Tale procedura è in grado di fornire informazioni sulla concentrazione di diversi fosfati, e del pH intracellulare dei muscoli, valori che possono essere successivamente utilizzati per determinare eventuali disfunzioni mitocondriali. I sintomi precoci dell’acidosi lattica sono: Nausea, vomito, diarrea e dolori addominali Mancanza di respiro e/o respiro corto Crampi, mialgia e parestesia
LCAD -
Mancanza A catena lunga Della Deidrogenasi Dell'Acilico-CoA Carenza di deidrogenasi Acyl-CoA a catena lunga. Sintomi: Di solito causa una sindrome fatale, negli infanti, tipicizzata da impossibilità a crescere, fegato dilatato, cuore allargato, encefalopatia metabolica, ed ipotonia. Sindrome di Leigh: da http://www.telethon.it/ Codice di esenzione: RF0030 Un gruppo di ricerca finanziato da Telethon, diretto dal Dott. Massimo Zeviani dell’Istituto nazionale Neurologico “C. Besta” di Milano, è giunto all’identificazione del gene responsabile della sindrome di Leigh, una gravissima malattia ereditaria mendeliana o sporadica, che colpisce oltre 1 individuo su 40.000. I bambini colpiti dalla sindrome già nei primi mesi di vita manifestano gravi sintomi di tipo neurologico, come incordinazione motoria, paralisi dello sguardo, difficoltà ad alimentarsi, e presentano un grave ritardo nello sviluppo. Presto la situazione si aggrava, e i piccoli muoiono entro i primi anni di vita. Da tempo è noto che questa terribile malattia è causata da alterazioni nel funzionamento dei mitocondri, i piccoli organuli che fungono da “centrale energetica” della cellula. Per lungo tempo, sospetti dei ricercatori si sono concentrati sul gene della citocromo ossidasi (o COX), un enzima-chiave nel funzionamento dei mitocondri, che in effetti risulta difettoso in almeno il 60 per cento dei bambini affetti dalla sindrome. Sorprendentemente, però, in nessuno dei pazienti si sono mai trovate anomalie nei geni della COX. Per risolvere questo
enigma i ricercatori milanesi hanno
utilizzato una tecnica recentissima che consiste nel trasferire singoli
cromosomi umani in cellule di pazienti per localizzare il
gene responsabile della malattia. E’ stato così possibile restringere il
campo ad un pugno di geni “indiziati”, situati in una regione del
cromosoma 9, e infine scoprire che uno di questi geni, chiamato SURF-1, è
alterato nella maggior parte dei pazienti analizzati. Eredità mendeliana recessiva. La malattia descritta per la prima volta dal dottor Leigh con ritardo dello sviluppo mentale e motorio (p.es. muscolo dormiente) dei neonati e dei bambini piccoli, frequenti attacchi epilettici; generalmente ereditato con ereditarietà diversificata. Ci sono anche malattie dei mitocondri, che in genere non comportano miopatia, per es. la neuropatia ottica di Leber, che causa gravi diminuzioni della vista per disturbi del nervo ottico. Attualmente si moltiplicano le indicazioni, che altre malattie neurodegenerative frequenti (con degenerazione delle cellule nervose) sono causate da difetti mitocondriali. Esemplare è l’atassia detta di Friedreich, nella quale sono colpiti il cervelletto ed il muscolo cardiaco. Non è ancora certo quale ruolo giochino i mitocondri nella demenza di Alzheimer, del morbo di Parkinson e della sclerosi laterale amiotrofica (SLA). La neuropatia ottica ereditaria di Leber (LHON) consiste in una disfunzione del nervo ottico dovuta a mutazioni nel DNA mitocondriale (mtDNA). L'eredità è non-mendeliana o materna; sono tuttavia numerosi i casi sporadici e isolati di LHON. La prevalenza è stimata in 1/50.000. La LHON si presenta spesso in soggetti giovani adulti, con un'età media d'esordio tra 18 e 35 anni. La perdita della vista inizia di solito in un occhio e può essere improvvisa, portando ad un'acuità inferiore a 20/400 in meno di una settimana, oppure può essere progressiva, nell'arco di 2-3 mesi. L'altro occhio può essere colpito contemporaneamente, in circa il 50% dei pazienti, oppure successivamente, con un intervallo fino a 9 mesi. L'esame del fondo dell'occhio rivela spesso pseudoedema del disco e iperemia, dilatazione arteriolare, tortuosità dei vasi e teleangiectasie peripapillari. Anche se la perdita della vista è di solito l'unico sintomo, sono state riportate associazioni della LHON con anomalie cardiache, neurologiche o scheletriche. L'atrofia ottica sembra essere legata alla disfunzione della catena respiratoria causata da mutazioni nel mtDNA. Nella LHON sono state osservate oltre 18 mutazioni nel mtDNA e almeno 4 corrispondono a "mutazioni primarie'', in quanto sono sufficienti a causare la malattia. Le principali mutazioni primarie del DNA mitocondriale coinvolgono i geni che codificano per diverse subunità dei complessi I e III della catena respiratoria mitocondriale. Altre mutazioni, conosciute come "mutazioni secondarie'', sono di solito associate a mutazioni primarie. Possono essere coinvolti nella patogenesi anche fattori epigenetici o tossici. Attualmente non esiste alcun trattamento efficace per la LHON
Malattia Di Luft Mancanza Della Carbossilasi Del Piruvato : Sintomi: Acidosi lattica, ipoglicemia, grave ritardo, mancata crescita. Sintomi comuni: Attacchi e spasticità. Mancanza Della Deidrogenasi Del Piruvato : Sintomi: Acidosi lattica, atassia, acidosi piruvica, degenerazione spinale e cerebrale. Meno comuni: Agenesi del corpo calloso e lesioni nel ganglio basale, cervelletto, e del tronco cerebrale. Anche: crescita ritardata, ipotonia, attacchi, e polineuropatia. Qualche volta si scopre che è la causa della Sindrome di Leigh Central core e minicore - Codice di esenzione: RFG070 - sinonimi
: - Miopatie Congenite Ereditarie
Molti lettori hanno contattato la nostra redazione per chiedere informazioni circa le malattie cosiddette central core e minicore (o multicore). Ce ne parla il professor Carlo Trevisan della Clinica neurologica II dell'Università di Padova. La Malattia "Central Core" è una miopatia congenita rara ad esordio precoce - non sono molti i casi di insorgenza in età adulta - che si caratterizza, all'esame istochimico del tessuto muscolare, per la presenza di aree prive di attività enzimatiche ossidative (i cosiddetti cores, termine inglese che letteralmente significa "noccioli"), al centro delle sezioni trasverse delle fibre di tipo 1 (quelle a contrazione lenta, spesso predominanti in questa patologia). Tali aree si producono per la mancanza di mitocondri. Sul piano clinico, il bambino, che alla nascita di solito presenta un lieve deficit del tono muscolare (ipotonia), avrà un ritardo dello sviluppo motorio, accompagnato da un deficit di forza variabile, ma per lo più non grave. Questa debolezza tende a rimanere stazionaria nel tempo anche se esiste la possibilità di una progressione lenta. Alla nascita possono essere presenti anche lussazione dell'anca e altre anomalie scheletriche. L'esame della CK sierica e l'elettromiografia sono generalmente normali o solo lievemente alterati. La malattia viene di solito trasmessa con modalità autosomica dominante: il gene è stato mappato sul locus q13.1 del cromosoma 19, come quello dell'Ipertermia maligna: questo rende ragione del fatto che i pazienti affetti da questa miopatia siano a rischio di incidenti anestesiologici anche mortali. La Malattia "Minicore (o "Multicore") è un'altra rara miopatia congenita solo in apparenza simile alla precedente. Le aree piccole e multifocali di degenerazione, caratteristiche di questa patologia, possono essere riscontrate nelle fibre muscolari di tipo 1 e 2 e si diffondono solitamente per la lunghezza di pochi sarcomeri lungo tali fibre. Esse presentano tratti caratteristici al microscopio elettronico, ma la loro presenza può anche essere aspecifica e secondaria ad altre miopatie. Di solito anche nella Malattia "Minicore" si riscontra una predominanza delle fibre di tipo 1. I sintomi si manifestano in genere nell'infanzia, con un'ipotonia lieve e non progressiva e debolezza dei muscoli delle braccia e delle cosce. Possono essere colpiti anche i muscoli extraoculari e facciali. La CK sierica risulta normale, mentre l'elettromiografia può essere di tipo miopatico. La modalità di trasmissione di questa patologia è diversa da quella della Malattia "Central Core": nella "Minicore", infatti, essa è soprattutto autosomica recessiva, anche se non può essere del tutto esclusa la trasmissione autosomica dominante. Anche questi pazienti, seppur in proporzione minore rispetto alla "Central Core", possono presentare suscettibilità all'ipertermia maligna, in corso di anestesia generale. Non esiste ancora purtroppo terapia farmacologica né per la malattia "Central Core", né per quella "Minicore". I pazienti possono trarre vantaggi da cicli di fisiokinesiterapia e da eventuali interventi ortopedici per le malformazioni ossee a volte associate.
Articolo tratto da DM 129 (febbraio 1998)
Malattia di Alzheimer: il cinque per cento circa dei malati anziani presenta la stessa mutazione del DNA mitocondriale Cancro: è recente la scoperta di una anomalia del DNA mitocondriale in una forma di tumore cerebrale ereditario molto aggressivo, il paraganglioma La sindrome da deplezione del DNA mitocondriale (MDS) comprende un gruppo clinicamente eterogeneo di malattie mitocondriali, caratterizzate da una riduzione del numero delle coppie del mtDNA nei tessuti affetti, senza mutazioni o riarrangiamenti nel mtDNA. Per questo la MDS si considera un processo primitivo. La deplezione del mtDNA può anche essere secondaria (come si avviene nella miosite da corpi inclusi) oppure iatrogena (come si riscontra nei pazienti trattati con analoghi nucleosidici). La MDS può essere una patologia neurogena relativamente frequente nel periodo neonatale e nell'infanzia. In una casistica, l'11% dei bambini (<2 anni), che presentavano debolezza, ipotonia e ritardo dello sviluppo, avevano una deplezione di mtDNA. La MDS è eterogenea a livello fenotipico e si può manifestare in forma epato-cerebrale, miopatica, miopatica benigna ad esordio tardivo o cardiomiopatica. I bambini di solito presentano nell'infanzia ipotonia, acidosi lattica e livelli elevati di creatin-chinasi sierica (CK). Alcuni di loro hanno anche una epatopatia grave, spesso fatale, oppure un coinvolgimento renale, che simula la sindrome di De Toni-Fanconi. La diagnosi clinica viene confermata dalla ridotta attività della catena respiratoria e, soprattutto, da un basso rapporto mtDNA/nDNA nei tessuti affetti. La MDS spesso ricorre nelle famiglie, con una possibile trasmissione autosomica recessiva, a sostegno di un difetto genetico del DNA nucleare. In effetti, le mutazioni nei geni della chinasi della deossiguanosina mitocondriale, codificata nel nucleo, e della chinasi della deossitimidina sono state associate rispettivamente alle forme epato-cerebrali e miopatiche della MDS. Questi due geni codificano enzimi per la via di salvataggio mitocondriale, implicata nella sintesi del mtDNA attraverso il rifornimento di deossiribonucelotidi (dNTPs). La debolezza muscolare e l'intolleranza all'esercizio possono rispondere all'integrazione con coenzima Q. *Autori: Dott. R. Carrozzo e S. Lucioli (Febbraio 2004).*
Principali manifestazioni cliniche delle malattie mitocondriali
Più che di epilessia è forse più corretto parlare di "epilessie". Si tratta di un gruppo di malattie neurologiche a carattere cronico, caratterizzate da crisi convulsive o da altre manifestazioni critiche di natura motoria, sensoriale, psichica e neurovegetativa, che hanno in comune la comparsa improvvisa e la ripetizione nel tempo. Una sola crisi non indica per forza che ci si trovi di fronte ad un caso di epilessia: infatti il 5% della popolazione ha avuto un attacco che si è verificato una sola volta in tutta la vita.
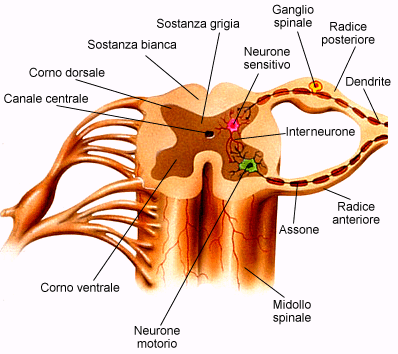
Il cervello è costituito da cellule nervose chiamate neuroni. La
proprietà fondamentale del tessuto nervoso è di emettere degli impulsi nervosi.
Questi sono costituiti da un debolissimo passaggio di corrente elettrica che si
genera nella cellula nervosa e si propaga ad altri neuroni con cui prende
contatto. Ogni funzione cerebrale (il movimento, il linguaggio, la vista, ecc…)
dipende da una perfetta e complicatissima regolazione della trasmissione degli
impulsi nervosi. Durante una crisi epilettica vi è un'improvvisa, abnorme e
sincronizzata scarica di impulsi nervosi da parte di un gruppo più o meno esteso
di cellule nervose. Questo è il meccanismo comune ad ogni tipo di crisi
epilettica; ciò che può cambiare sono le manifestazioni cliniche, cioè i segni
visibili delle crisi. I meccanismi di base che entrano in gioco sono molto
complessi ed ancora oggi non del tutto chiariti.
Definizione Paralisi dei muscoli oculari. Può essere:
Cause: Lesione di natura nervosa o muscolare.
L'oftalmoplegia esterna progressiva è
una forma clinica benigna caratterizzata da oftalmoparesi, ptosi
palpebrale(abbassamento
della palpebra superiore, destra o sinistrae),
spesso, debolezza muscolare, prevalentemente cingolare. L'esordio è
nell'adolescenza o nell'età adulta, l'evoluzione è lenta e progressiva. Nel 50
per cento dei pazienti la malattia è causata da una delezione del DNA
mitocondriale (casi sporadici), mentre in un terzo si tratta di una mutazione
puntiforme (casi a trasmissione materna non mendeliana).
deviazione patologica di
uno o di entrambi gli occhi, i rapporti dei cui assi risultano alterati, così da
produrre una duplicità dell’immagine osservata. Molto comune è lo strabismo
latente (o eteroforia), che risulta evidente solo quando si esauriscono i
riflessi di compensazione che l’individuo mette inconsciamente in opera. Nello
strabismo vero (o eterotropia), si hanno deviazione degli assi visivi e
alterazioni della visione binoculare, senza alterazioni dei movimenti oculari
(strabismo concomitante); può essere unilaterale, alternante, periodico; inoltre
l’asse dell’occhio colpito può divergere (extropia), convergere (esotropia),
elevarsi (ipertropia) o risultare abbassato (strabismo deorsum vergens)
rispetto all’altro occhio. Lo strabismo vero può dipendere da disturbi dei
centri dello sguardo e dell’innervazione dei muscoli estrinseci degli occhi, da
difetti degli stessi muscoli, da alterazioni del globo oculare e dei suoi mezzi
diottrici. Lo strabismo paralitico si verifica in caso di paralisi di uno o più
muscoli estrinseci oculari originate da lesioni nervose centrali o periferiche;
è questo il tipo di strabismo che più frequentemente dà luogo a diplopia, perché
nelle altre forme l’individuo si abitua solitamente a escludere dalla visione
l’occhio colpito, così da percepire una sola immagine ( ambliopia). È
caratterizzato da deviazione degli assi visivi con alterazione di alcuni
movimenti oculari, diversi a seconda del muscolo colpito.
riduzione in peso e volume di un organo;
in genere è dovuta ad alterazioni nei processi di Biosintesi(sintesi di composti
chimici effettuata da organismi viventi o comunque realizzata nell’interno di
essi. La capacità di biosìntesi è un requisito fondamentale della materia
vivente che garantisce la formazione e il ricambio dei componenti cellulari ,
nonché un regolare svolgimento degli scambi energetici) facile stancabilità all'esercizio fisico Stanchezza molto forte, sia fisica che mentale. Col caldo, la stanchezza diminuisce un poco, mentre col freddo aumenta. Deterioramento della muscolatura, che si manifesta con debolezza e intolleranza al movimento; i muscoli mostrano spesso fibre rosse sfilacciate, piene di mitocondri anormali, che diventano rossi quando sono in presenza di un particolare colorante.
termine con cui si
indicano genericamente tutte le patologie che interessano i muscoli.
Sotto questo termine sono raggruppate molteplici
malattie che colpiscono l'unità motoria. L'unità motoria comprende il neurone
motorio, localizzato nel midollo spinale, il suo prolungamento o assone nel
nervo periferico, la giuntura neuromuscolare e l'insieme delle fibre muscolari
innervate dal neurone. L'unità motoria è l'unità anatomica e funzionale
terminale del sistema motorio. Tutte le malattie dell'unità motoria si
manifestano con un deficit motorio (paresi o debolezza muscolare; plegia o
paralisi). Il deficit varia secondo la gravità e la localizzazione
(generalizzata o localizzata al viso, al tronco, alle membra), secondo la
malattia.
Disturbo della coordinazione del corpo in rapporto
con l’equilibro, sia nel mantenimento della postura (atassìa statica), sia nella
marcia (atassìa dinamica). L’atassìa può derivare da lesioni cerebellari, da
disturbi della sensibilità profonda (tabe, polineuropatie, lesioni del lobo
parietale), da disturbi labirintici o da lesioni cerebrali (del lobo frontale,
temporale o del corpo calloso). La prova di Romberg permette di cogliere anche
modeste turbe atassiche: il soggetto viene posto sull’attenti, con le punte dei
piedi unite, e quindi invitato a chiudere gli occhi. Se, a occhi chiusi,
oscilla, fino a tendere a cadere, la prova indica lesioni dei cordoni posteriori
o malattie del labirinto dell’orecchio. Il malato con lesioni cerebellari,
invece, oscilla già a occhi aperti, e l’equilibrio non viene peggiorato dalla
chiusura degli occhi: in questo caso, la prova di Romberg è negativa. Termine generico usato per indicare movimenti anormali, involontari, causati da disturbi funzionali di un dato distretto del sistema nervoso. Si tratta di movimenti che si aggiungono a quelli volontari normali disturbandoli; sono diversi fra loro per qualità, distribuzione e intensità: in alcuni casi si osservano semplici contrazioni di muscoli isolati, in altri i sintomi sono più complessi. Le coree, i crampi, il
morbo di Parkinson, gli spasmi, il tremore, il
tic, il riso e il pianto spasmodici sono tipici esempi di discinesia. Sono
discinesìe anche alcune particolari condizioni di alterata motilità di organi,
con sintomi dolorosi, che colpiscono il tubo gastroenterico, la colecisti, le
vie biliari e le vie urinarie. Una forma particolare è la discinesia tardiva,
che si manifesta in seguito a terapie prolungate con farmaci antidepressivi.
Termine generico usato per
indicare movimenti anormali, involontari, causati da disturbi funzionali di un
dato distretto del sistema nervoso. Si tratta di movimenti che si aggiungono a
quelli volontari normali disturbandoli; sono diversi fra loro per qualità,
distribuzione e intensità: in alcuni casi si osservano semplici contrazioni di
muscoli isolati, in altri i sintomi sono più complessi. Le
coree,
i crampi, il
morbo di Parkinson,
gli spasmi, il tremore, il tic, il riso e il pianto spasmodici sono tipici
esempi di discinesia. Sono discinesìe anche alcune particolari condizioni di
alterata motilità di organi, con sintomi dolorosi, che colpiscono il tubo
gastroenterico, la colecisti, le vie biliari e le vie urinarie. Una forma
particolare è la discinesia tardiva, che si manifesta in seguito a terapie
prolungate con farmaci antidepressivi.
sindrome caratterizzata da
tipici movimenti involontari, improvvisi, irregolari, scattanti, presenti a
riposo e nell’esecuzione di atti volontari, accentuati dalle emozioni, dalla
fatica e dal freddo, assenti nel sonno (movimenti coreici). Tali movimenti,
variamente associati a ipotonia muscolare e a turbe psichiche, sono il sintomo
tipico di alcune malattie del sistema extrapiramidale (còrea di Sydenham e còrea
di Huntington). Possono essere presenti pure in caso di lesioni di strutture
extrapiramidali, dovute a encefaliti, trombosi, emorragie, tumori (còree
sintomatiche). Per la còrea di Sydenham, vedi ballo di san vito. La còrea di
Huntington, o còrea degenerativa, è una malattia gravissima, ereditaria, che
insorge in età adulta. È dovuta ad atrofia e degenerazione dei gangli della base
e della corteccia cerebrale, con sbilanciamento nell’equilibrio dei
neurotrasmettitori a favore della dopamina. È caratterizzata dai movimenti
coreici e da turbe psichiche, che iniziano insidiosamente con alterazioni del
carattere e del comportamento, per poi evolvere in demenza, con frequente
tendenza al suicidio e all’abuso di bevande alcoliche. I farmaci neurolettici
sono utili contro i grandi movimenti coreici. Fondamentale è la prevenzione a
livello di consulenza genetica (si sconsiglia di avere figli a un membro di una
famiglia in cui si ripetono casi di còrea di Huntington). Da segnalare la còrea
delle gravide: rara, insorge quasi esclusivamente nelle primipare durante il
purperio, è grave, con sintomi simili al ballo di san Vito. Contrazione brusca e involontaria che può interessare pochi o numerosi gruppi muscolari, a carattere irregolare, intermittente e variabile. Mioclonìe: Può interessare: uno o pochi fascicoli muscolari, senza dar luogo a movimenti; un muscolo isolato (mioclonìa elementare); i muscoli sinergici di uno o più arti, con spostamento di segmenti corporei (mioclonìe segmentarie); oppure tutto il corpo (mioclonìe massive o generalizzate, o scosse miocloniche). Le mioclonìe possono essere spontanee o provocate da stimoli vari, intermittenti o permanenti. Mioclonìe elementari si riscontrano fisiologicamente in tutti i soggetti normali durante il sonno. Contrazione clonica(contrarsi e rilassarsi di un muscolo caricato al limite delle capacità) di uno o più muscoli, con sede più frequente negli arti o nel tronco, rapida, talora accompagnata da tremori fibrillari, con effetto locomotore limitato o nullo, non ritmica a frequenza variabile. L'emozione , il freddo e l'eccitazione meccaniche ne aumentano l'intesità. ictus o apoplessia l’ictus cerebrale ischemico, vale a dire l’ostruzione di un’arteria cerebrale, un incidente che può portare a gravi invalidità permanenti. Complesso di sintomi caratterizzati da ictus celebrale con blocchi delle funzioni celebrali più importanti, perdita di coscienza, della parola e delle funzioni sensoriali e motorie. La gravità dipende dal vaso interessato, dalla consistenza dell'ostruzione e dalla regione celebrale implicata. neuropatia ottica
COS'È UNA NEUROPATIA PERIFERICA? Il sistema nervoso consiste di due componenti: il sistema nervoso centrale, costituito da encefalo e midollo spinale; e il sistema nervoso periferico, costituito da nervi che connettono il sistema nervoso centrale ai muscoli, alla pelle e agli organi interni. "Periferico" significa "lontano dal centro", proprio ad indicare la funzione che i nervi hanno di collegare il sistema nervoso centrale agli organi periferici. Il sistema nervoso periferico è la parte danneggiata in corso di neuropatia. Neuropatia è, quindi, il termine usato per descrivere disturbi conseguenti a danno dei nervi periferici. COME É FATTO UN NERVO? L'unità base del sistema nervoso periferico è il "neurone" o cellula nervosa. Suo compito è inviare informazioni da una parte all'altra del corpo attraverso impulsi elettrici. Ciascun nervo è formato da un corpo cellulare e da un lungo prolungamento chiamato "assone". L'assone conduce impulsi tra il corpo cellulare e la periferia, dove entra in contatto con strutture specializzate (recettori), presenti nei muscoli, nella pelle e negli organi interni. Molti assoni sono avvolti da una membrana, chiamata "guaina mielinica", che permette agli impulsi elettrici di trasmettersi in maniera più veloce ed efficiente. Gli assoni viaggiano insieme uniti in tronchi nervosi, che spaziano per il corpo umano come "cavi" di un complesso "circuito elettrico". TRE TIPI DI NERVI
In base al tipo di fibre che contengono, si possono identificare
tre tipi di nervi: motori, sensitivi, e vegetativi o autonomici. È UNA MONO- O UNA POLINEUROPATIA?
La malattia di un singolo nervo periferico si chiama
mononeuropatia. Le mononeuropatie colpiscono singoli nervi in areee ben definite
e spesso sono conseguenza di una lesione traumatica, di una compressione locale
(con "schiacciamento" del nervo) o di processi infiammatori o ischemici. La
sintomatologia è, pertanto, localizzata e limitata al territorio di innervazione
del nervo leso. Esempi di mononeuropatie sono la sindrome del tunnel carpale, e
la paralisi di Bell. La sindrome del tunnel carpale consegue a una compressione
del nervo mediano nel suo passaggio a livello del polso. La compressione del
nervo può essere secondaria a uso eccessivo del polso o a processi infiammatori;
talora sono presenti condizioni sottostanti quali diabete, artrite reumatoide,
acromegalia. QUALI SONO I SINTOMI? Alcune neuropatie esordiscono in maniera improvvisa, altre in maniera graduale nell'arco di anni. I sintomi dipendono dal tipo di fibre nervose interessate (motorie, sensitive, vegetative) e dalla loro localizzazione, ma nella maggior parte dei casi si manifestano con debolezza, formicolìì e dolore, come elencato brevemente di seguito. Debolezza alle braccia o alle gambe. La debolezza muscolare, l'astenìa sono sintomi dovuti a una compromisione dei nervi motori. Se sono interessati gli arti inferiori, si possono manifestare facile affaticabilità e senso di "pesantezza" alle gambe, difficoltà nel salire le scale, nel camminare o correre. Se sono interessati gli arti superiori, si può provare fatica nel portare la borsa della spesa, nello svitare i coperchi dei barattoli, nell'aprire la porta, o nel pettinarsi. Intorpidimento, formicolìo, dolore Una lesione dei nervi sensitivi può causare sintomi molto diversi. Possono esserci sensazioni spontanee (parestesìe), che includono intorpidimento, formicolìì, sensazione di "spilli" o aghi o pizzicotti, prurito, bruciori, freddo, fitte dolorose e profonde, scosse elettriche. Questo disturbi spesso peggiorano di notte. Si possono, inoltre, avere sensazioni spiacevoli scatenate dallo stimolo tattile (disestesie), oppure riduzione (ipoestesia) e scomparsa (anestesia) della sensazione, che possono far sì che ci si tagli o scotti senza rendersene conto. Assenza del senso di posizione In presenza di questo disturbo, non si è sicuri di dove si trovino esattamente i piedi e può insorgere incoordinazione e insicurezza nel camminare. Oppure ci si può accorgere che il modo di camminare si è modificato, senza capire esattamente come o perchè. È possibile "trascinare" i piedi, oppure la marcia si allarga nel tentativo inconscio di mantenere l'equilibrio. Sensazione di "guanti" e "calzini" È la sensazione di stare indossando guanti, calzini o ciabatte, quando invece mani e piedi sono completamente nudi. Sintomi di danno alle fibre autonomiche Una lesione delle fibre autonomiche può causare senso di instabilità e/o vertigini quando si è in piedi, costipazione, diarrea, disfunzioni sessuali, e assottigliamento della pelle, con facilità a sviluppare lividi e difficoltà nella guarigione delle ferite.
BREVE GUIDA ALLE CAUSE DI NEUROPATIA
Si possono distinguere due grossi gruppi di neuropatie:
ereditarie (causate da anomalie genetiche) e acquisite (dovute, cioè, a malattie
acquisite nel corso della vita). La maggior parte delle neuropatie sono
acquisite, e possono essere dovute a diverse cause. Quando la causa della
neuropatia non è nota, si parla di neuropatie "idiopatiche".
NEUROPATIE ACQUISITE
1) Diabete
2) Alcool e altre sostanze tossiche
3) Deficit nutrizionali (neuropatie carenziali)
4) Neuropatie in corso di malattie sistemiche
5) Neuropatie immuno-mediate
a. Sindrome di Guillain-Barrè
b. Poliradicolonevrite infiammatoria demielinizzante cronica
c. Neuropatie croniche con autoanticorpi verso i
nervi periferici
d. Neuropatie associate a vasculiti
e. Neuropatie associate a gammopatie monoclonali
6) Tumori
7) Amiloidosi
8) Agenti infettivi
9) Farmaci
10) Trauma o compressione
11) Idiopatiche NEUROPATIE EREDITARIE Le neuropatie ereditarie sono causate da alterazioni genetiche che vengono trasmesse di generazione in generazione. Per molte di queste il difetto genetico è noto e sono disponibili tests diagnostici.
1) HSMN (Hereditary Sensory Motor Neuropathy) o Malattia di
Charcot-Marie-Tooth (CMT)
Neuropatia amilodotica familiare
Neuropatie in corso di porfiria DIAGNOSI
Una corretta diagnosi inizia da una raccolta dettagliata dei
sintomi, delle malattie pregresse e concomitanti del paziente, da una ricerca di
possibili fattori causali (esposizione a tossici, farmaci, etc.). Dopo aver
attentamente visitato il paziente, il neurologo consiglia alcuni esami di
laboratorio che possono aiutare a identificare la causa della neuropatia. COME SI TRATTA UNA NEUROPATIA?
Gli obbiettivi del trattamento sono due:
1) Eliminare la causa della neuropatia
2) Ridurre i sintomi della neuropatia COME PUOI AIUTARTI DA SOLO, GIÀ FIN D'ORA
In presenza di sintomi simili a quelli descritti in queste
pagine, non aspettare a vedere cosa succede. È possibile, infatti, che tu sia
affetto da una neuropatia cronica, che non guarirà da sola, al contrario
peggiorerà col tempo e sarà poi più difficile trattare.
C'è da puntualizzare che la Malattia del Motoneurone (MND), non coincide con la SLA, in quanto quest'ultima è compresa nel più ampio gruppo delle MND, che annovera anche la Sclerosi Laterale Primaria (Primary Lateral Sclerosis, PLS), LAtrofia Muscolare Spinale Progressiva (Progressive Spinal Muscular Atrophy, PSMA), le Atrofie Muscolari Spinali segmentali, focali e distali, la Neuropatia Motoria Multifocale (Multifocal Motor Neuropathy, MMN), l'Atrofia Muscolare Bulbospinale Progressiva o Malattia di Kennedy (Progressive ulbospinal Muscular Atrophy, PBSMA), la Sindrome di Brown-Vialetto-Van Laere (Paralisi pontobulbare con sordità sensoriale), l'Atrofia Muscolare Spinale (Spinal Muscolar Atrophy, SMA) correlata con mutazioni del gene smn e la SMA familiare progressiva a esordio tardivo, la Paraplegia Spastica Ereditaria o Malattia di Strùmpel-Lorrain (Hereditary Spastic Paraplegia, HSP), la Sindrome di Fazio-Londe(Paralisi infantile bulbare progressiva), e altre forme patologiche e eziopatogenesi tossica, attinica o infettiva - come la Sindrome PostPolio, che si verifica in soggetti con pregressa poliomielite - : clinicamente, peraltro, i termini SLA e MND vengono considerati sinonimi, a indicare una patologia rapidamente progressiva caratterizzata da paralisi, ipertrofia, atrofia muscolare, fascicolazioni e crampi.
sordità neurale Menomazione uditiva mitocondriale I Potenziali evocati I potenziali evocati rappresentano la risposta a livello cerebrale o periferico ad uno stimolo sensoriale. Normalmente tale risposta è di bassa ampiezza e si confonde con l'attività di fondo. Tuttavia, se si fa la media delle risposte ad un numero sufficiente di stimoli tutti uguali, è possibile far emergere la risposta strettamente correlata a quel dato stimolo. Lo scopo di tale tipo di indagine è quello di verificare l'integrità di funzionamento della particolare via sensoriale indagata. Esistono quindi specifici esami per le varie modalità sensoriali:
e nel registrare la risposta dalle regioni occipitali con elettrodi di superficie tipo EEG. Tempo di esecuzione: circa 30 min. Nota Ben qualora il soggetto faccia uso di occhiali è necessario che li porti.
con rumore bianco e registrando con elettrodi di superficie dalla sommità del capo. Servono prevalentemente ad esplorare le vie acustiche nel loro decorso lungo il tronco dell'encefalo. Importanti riduzioni dell'udito ne compromettono l'attendibilità. Tempo di esecuzione: circa 40 min.
al polso (per gli arti superiori) o alla caviglia (per gli arti inferiori) e registrando l'impulso sensitivo lungo il decorso del nervo, a livello vertebrale e cerebrale con elettrodi di superficie. Poiché l'esame può risultare talora un po' fastidioso ed è per contro necessario che il soggetto sia rilassato, viene spesso somministrata prima una bassa dose di ansiolitico. Per tale motivo si sconsiglia di porsi alla guida subito dopo l'esame. Tempo di esecuzione: circa 60 min. (per coppia di arti).
a livello sacrale. Tempo di esecuzione: circa 45 min.
vengono intercalati ad altri stimoli molto più frequenti ma con caratteristiche diverse e registrando una specifica risposta di "riconoscimento" che si genera a livello cerebrale. Vengono utilizzati nella valutazione delle demenze. Tempo di esecuzione: circa 45 min.
rispettivamente la corteccia cerebrale o il midollo spinale. Lo stimolo induce una risposta motoria che viene registrata ad alcuni muscoli degli arti superiori ed inferiori con elettrodi di superficie, consentendo di valutare la funzionalità delle vie motorie. La stimolazione non è particolarmente fastidiosa ed è normalmente ben tollerata. Tempo di esecuzione: circa 60 min. Nota Bene: la stimolazione magnetica non può essere eseguita in pazienti con dispositivi impiantati meccanici od elettrici (es. pace-maker) né in pazienti con oggetti o frammenti metallici magnetizzabili nel capo (es. clips da intervento).
"La demenza consiste nella compromissione globale delle funzioni cosiddette corticali (o nervose) superiori, ivi compresa la memoria, la capacità di far fronte alle richieste del quotidiano e di svolgere le prestazioni percettive e motorie già acquisite in precedenza, di mantenere un comportamento sociale adeguato alle circostanze e di controllare le proprie reazioni emotive: tutto ciò in assenza di compromissione dello stato di vigilanza. La condizione è spesso irreversibile e progressiva." Mentre nella maggior parte delle patologie neurologiche i sintomi cognitivi sono la diretta conseguenza di una lesione immediatamente osservabile (ad esempio attraverso specifici esami come la Tac o una Risonanza Magnetica), l’esordio di alcune forme di demenza è caratterizzato da deficit cognitivi che si manifestano isolatamente, cioè senza un preciso riscontro neurologico. Il punto cruciale della diagnosi, allora, consiste nel verificare se questi disturbi siano da considerarsi patologici o conseguenza del normale declino cognitivo legato all’età, al livello culturale del soggetto e alle sue condizioni generali di vita. Per fare ciò i familiari non devono accettare come unica spiegazione il fatto che la persona stia semplicemente invecchiando. In altre parole, è necessario che certi comportamenti insoliti, ovviamente se osservati con una certa frequenza, conducano alla consultazione dello specialista (vedi riquadro 4). In queste situazioni un accurato Esame Neuropsicologico consentirà di stabilire se i fenomeni osservati siano conseguenza di una patologia neurologica in atto, il frutto di un particolare momento di vita caratterizzato da aspetti ansiosi o depressivi, oppure la normale conseguenza dell’invecchiamento cerebrale. Il processo diagnostico richiede la collaborazione dello specialista neuropsicologo e del medico neurologo e del geriatra, fondamentale laddove si tratti di un paziente in terza età. Ovviamente è buona prassi intrecciare un rapporto di collaborazione anche con il medico curante, sicuramente il clinico che meglio conosce il paziente e le sue abitudini di vita. Nel caso in cui si dovesse giungere alla diagnosi di demenza, è necessario attivare subito tutte le risorse necessarie per fronteggiare il problema.
Con il termine cefalea si intende il dolore al capo
presente in molte malattie febbrili, infettive e nervose, oppure presente di per
sé, come sintomo isolato. Il mal di testa è probabilmente uno dei disturbi più
comuni , ma spesso rappresenta un problema sia per il medico che per il
paziente, perché generalmente è un sintomo provocato da innumerevoli
disfunzioni. Le cause possono essere ricercate in malattie degli occhi, del
naso, delle orecchie, delle strutture intracraniche che coinvolgono il cervello.
Sovente l'origine della cefalea sono disturbi
emotivi o lo stress, oppure malattie generali dell'organismo, accompagnate da
febbre. Esiste probabilmente anche una predisposizione ereditaria nei confronti
del mal di testa. (
http://www.cefalea.it ) Mancanza di tonicità di qualsiasi organo o tessuto. Alterazione del tono muscolare. Alterazione dell’equilibrio fra il tono simpatico(sezione del sistema nervoso autonomo costituita da centri situati nel tratto toraco-lombare del midollo spinale) e il tono parasimpatico[componente del sistema nervoso autonomo vegetativo che, insieme al sistema simpatico, regola le funzioni dei singoli organi e l’ omeostasi( capacità da parte dell'organismo di mantenere un equilibrio interno stabile, grazie a un insieme di processi di regolazione e controregolazione che agiscono ogniqualvolta si verifica una variazione delle condizioni esterne) dell’intero organismo], con prevalenza del primo (simpaticotonia) o del secondo (vagotonia). I disturbi funzionali che la caratterizzano dipendono dall’iperattività dei due sistemi:tachicardia, pallore, ipertensione nel primo caso; bradicardia, secchezza orale, turbe gastroenteriche, midriasi nel secondo. ARD, Associazione italiana per la ricerca sulla distonia: http://www.distonia.it
Alterato sviluppo del miocardio con ipertrofia globale, predominante a carico del ventricolo sinistro, con frequenti alterazioni del ritmo e ad evoluzione rapidamente progressiva verso l'insufficienza cardiaca.
La cardiomiopatia ipertrofica (CMI) è causalmente
legata a difetti nei geni delle proteine dei sarcomeri e, meno frequentemente,
nei geni del DNA mitocondriale (mtDNA). La malattia
può essere asintomatica, aritmogenica o complicata da scompenso cardiaco
congestizio. Mutazioni, presenti nei membri affetti di una famiglia, spiegano
solo in parte i loro fenotipi e geni modificatori nucleari sono stati indicati
avere un ruolo nell'espressione dell'ipertrofia. Per ora, nulla a questo
riguardo, è stato osservato per l'mtDNA.
difetti della conduzione cardiaca Alterazioni del ritmo cardiaco da compromissione della diffusione dello stimolo elettrico di eccitamento; può essere asintomatico o manifestarsi in forme diverse: bradi- o tachi-cardia, blocchi più o meno completi, aritmie. retinopatia pigmentaria - codice di esenzione RFG110 - Con il termine Retinite Pigmentosa (RP) si definisce un gruppo di malattie caratterizzate da una degenerazione progressiva della retina che interessa entrambi gli occhi. Spesso accanto al termine Retinite Pigmentosa vengono utilizzate anche altre denominazioni dal significato analogo come: Retinosi Pigmentaria, Retinopatia Pigmentosa, Degenerazione Tapeto-Retinica, ecc.
La Retinite Pigmentosa è una malattia degenerativa
che colpisce le cellule fotorecettrici della retina (i bastoncelli ed i coni)
uccidendole lentamente. In questo modo la capacità visiva del soggetto colpito
Le cause che determinano questa infermità sono
ancora sconosciute e di conseguenza non esiste alcuna cura per i malati.
L’unica informazione di cui gli scienziati
dispongono è che la Retinite Pigmentosa, viene trasmessa ereditariamente, di
generazione in generazione, seguendo meccanismi ormai noti ai genetisti. Nel caso di matrimonio, una persona colpita da Retinite Pigmentosa dovrebbe ricorrere ad approfonditi esami genetici per poter quantificare i rischi di trasmissione della malattia.
Restringimento e/o degrado centrale del campo visivo
Il decorso della malattia ha una durata estremamente
variabile ma è comunque quasi sempre progressivo.
Opacità del cristallino. Può essere senile (più frequente), congenita (spesso associata ad altre malformazioni o a una infezione congenita come la rosolia), traumatica, tossica (dinitrofenolo, corticoterapia protratta), da malattie metaboliche (diabete mellito, galattosemia, ipoparatiroidismo). L'opacità può interessare la capsula (c. capsulare), la lente del cristallino (c. lenticolare) o entrambe (c. capsulo-lenticolare).
Può essere conseguente ad un aumento della produzione di acidi nel sangue, ma anche dalla perdita di bicarbonati a causa di una grave diarrea. Il metabolismo corporeo produce acido (carbonico) respiratorio. L'insufficienza cardiorespiratoria tende a produrre un accumulo di acido lattico, metabolico. Perciò in emergenza è usuale trovare che il trattamento richiesto consiste nella correzione di un acidosi metabolica o respiratoria. Per questa ragione, e per semplificare, nei paragrafi seguenti si tratta principalmente dell'acidosi e della sua correzione:
Acido lattico:composto
organico derivante dal metabolismo dei carboidrati. Il glucosio metabolizzato
nei tessuti poveri di ossigeno e nei muscoli durante il lavoro intenso o
prolungato, una volta ridotto a acido piruvico, non può essere ossidato fino ad
anidride carbonica e acqua, per cui si ha la riduzione dell’acido piruvico a
àcido làttico mediante una reazione catalizzata dalla latticodeidrogenasi.
Quando la concentrazione di latticodeidrogenasi nel sangue aumenta (per esempio,
nel corso del lavoro muscolare intenso) intervengono vari meccanismi che tendono
a rimuovere l’eccesso di àcido làttico: una parte viene utilizzata nel fegato
per risintetizzare glicogeno, una parte viene eliminata come tale con le urine,
una parte subisce la demolizione totale ad anidride carbonica e acqua.
Quest’ultimo processo avviene con consumo di ossigeno e comporta la produzione
di una certa quantità di ATP e di energia.
Glucosio:monosaccaride
molto diffuso in natura, in forma semplice oppure in molecole complesse
costituite da molte unità concatenate (come gli amidi, il glicogeno, la
cellulosa). È il carboidrato più utilizzato dai tessuti dell’organismo. Il
glucosio è contenuto allo stato libero in numerosi frutti. Esso è presente nel
sangue in concentrazioni che variano fisiologicamente nell'arco delle 24 ore;
non è pertanto possibile indicare un valore assoluto di normalità per la
glicemia: per questo motivo, è più opportuno fare riferimento a diversi valori
di normalità della glicemia, a digiuno o post-prandiale. Il suo potere
dolcificante è circa la metà di quello del saccarosio. I disaccaridi
(saccarosio, lattosio, maltosio) e i polisaccaridi (amido, glicogeno), che
contengono il glucosio come componente essenziale, vengono scissi in glucosio
prima di essere assorbiti nel tubo digerente; dopo essere stato assorbito il
glucosio giunge, attraverso la vena porta, al fegato. Il fegato in genere
trasforma anche gli altri monosaccaridi, assunti con la dieta, in glucosio;
tutto il glucosio è poi ceduto dal fegato al sangue e quindi agli altri tessuti
in misura rispondente alle esigenze energetiche, oppure accumulato sotto forma
di glicogeno. ATP:sigla dell’acido adenosintrifosforico, sostanza importantissima per il metabolismo degli organismi viventi, quale accumulatore di energia; si forma durante la distruzione delle molecole di alimento e si scinde facilmente in acido adenosindifosforico (ADP) e acido fosforico, sviluppando una grande quantità di energia che serve per tutti i processi vitali. I processi ossidativi (respirazione) servono a rigenerare ATP da ADP e acido fosforico. Nelle piante verdi l’ATP si origina per via fotochimica (vedi anche energia: consumo e produzione ).
Termine generico per indicare una malattia del fegato. Termine che sta ad indicare una condizione di compromissione della funzionalità renale. In questo gruppo sono comprese quelle nefropatie nelle quali la disfunzione renale insorge secondariamente ad altre patologie. pseudo-obstruzione intestinale I disordini gastroenterici, determinati dall'alterazione del controllo della motilità viscerale nelle mitocondriopatie. I disturbi digestivi risultano subdoli, poiché possono decorrere inosservati come sensazioni di fastidiosa ripienezza postprandiale, lievi dolori addominali, episodi di vomito, diarrea e/o stipsi.
Notevole riduzione del numero degli elementi cellulari del sangue
circolante (eritrociti, leucociti e piastrine). Le piastrine sono costituite da una parte centrale più rifrangente (cromomero) e da una parte più pallida (ialomero). Svolgono funzioni di grande importanza per l'emostasi e per la coagulazione del sangue: infatti si addensano e si conglutinano nei punti dove si verificano lesioni dei vasi e, dissolvendosi, liberano le sostanze che partecipano alla coagulazione del sangue. Le piastrine hanno una vita media di nove giorni, e il loro numero è normalmente di 200 000-300 000 per ml di sangue. La milza e il fegato sono gli organi principalmente deputati alla rimozione delle piastrine alterate o senescenti. Con questo termine viene indicato un gruppo di anemie caratterizzate dall'aumento del numero di sideroblasti nel midollo osseo e dalla loro ricchezza in ferro. I sideroblasti sono eritroblasti che contengono granulazioni di ferro non emoglobinico, visibile dopo colorazione con blu di Prussia. L'utilizzazione del ferro nella sintesi della emoglobina è alterata, per cui si ha una anemia ipocromica.
Eritroblasti:
precursore del globulo rosso, ancora provvisto di nucleo, presente nel midollo
osseo. È la prima cellula della serie rossa capace di sintetizzare l’emoglobina.
L’eritroblasto, dopo varie divisioni e maturazioni, espelle il nucleo e si
trasforma in reticolocito. L’eme è in grado di legare l’ossigeno reversibilmente; è responsabile inoltre del colore rosso diabete melitto: all'1,5 per cento dei casi viene attribuita a una mitocondriopatia. Il diabete è una malattia metabolica cronica, in cui l’organismo, non essendo in grado di produrre o utilizzare completamente l’insulina, causa un difetto del metabolismo dei carboidrati e, in misura minore, delle proteine e dei grassi, per cui i reni, per liberare l’organismo dall’eccesso di zuccheri (glicemia)cominciano a eliminarli attraverso l’urina. L’aumento della glicemia provoca danni in tutte le strutture dell’organismo, aggravando il rischio di complicanze, quali disturbi circolatori, insufficienza renale,ictus, e problemi neurologici. Vi sono due principali tipi di diabete: il tipo I, o diabete mellito insulino-dipendente in cui la produzione di insulina cessa completamente e il diabete di tipo II, in cui l’organismo non produce una quantità sufficiente di insulina o non è in grado di utilizzarla. Il diabete mellito di tipo I è detto anche diabete giovanile poiché colpisce i bambini e i giovani e solitamente si manifesta entro i 20 anni, quando le cellule insulari pancreatiche vengono distrutte, compromettendo la produzione di insulina. Esso costituisce il 10-15% dei casi di diabete mellito. Il diabete mellito di tipo II, o non insulino-dipendente, detto anche diabete senile ha uno sviluppo più lento e colpisce in genere le persone anziane sovrappeso ma può manifestarsi anche in soggetti di peso normale. La sintomatologia del diabete mellito, pur manifestandosi con caratteristiche ben determinate, presenta una grande varietà di espressioni legate sia al grado di gravità della malattia sia a condizioni oggettive quali l’età del paziente, la concomitanza di altre malattie, ecc. Viene diagnosticato attraverso l'individuazione di presenza di glucosio nell’urina e livelli anomali di glucosio nel sangue. La diagnosi è risolta dopo restituzione alla normalità di tali parametri per trattamento insulinico. L’insorgere del diabete può manifestarsi in modo molto poco avvertibile. Una maggiore affaticabilità, la comparsa di prurito specialmente in regione anale e vulvare, un aumento della quantità delle urine, una sete non giustificata, frequenti infezioni della pelle e lenta cicatrizzazione delle ferite, dovrebbero consigliare un esame delle urine, alla ricerca dell’eventuale presenza di glucosio. A uno stadio più avanzato, segni che devono far sospettare il diabete mellito sono: dimagramento, disturbi digestivi, nausea, nevralgie, turbe della vista, crescente affaticabilità, aumento dell’urina prodotta rispetto all’acqua ingerita. Molto importanti sono i segni che denunciano un’alterata circolazione specialmente agli arti inferiori (chiazze livide sulla cute, dolori, alterazioni della sensibilità, ecc.) e la possibilità di una gangrena. A livello dell’apparato genitale importanti sono i segni di impotenza sessuale nel maschio e di alterazioni del ciclo mestruale nella donna. I danni al sistema nervoso si manifestano con intorpidimento intellettuale e depressione. Nel diabete giovanile, la sintomatologia insorge in modo brusco e si sviluppa più rapidamente che nell’adulto. Il diabete mellito colpisce all'incirca l'1-2% della popolazione. Può provocare danni a cuore, occhi, reni e arti, e costituire un fattore di rischio nel caso di una eventuale gravidanza. Un adeguato e tempestivo trattamento può, tuttavia, ridurre notevolmente queste complicanze. Glicemia: L'esame determina il valore della concentrazione di glucosio presente nel sangue. Il glucosio deriva principalmente dalla alimentazione, oltre che dalla glicogenolisi epatica e dalla gliconeogenesi da parte del fegato di precursorsi non glicidici. A cosa serve: Serve ad individuare il rischio di ammalarsi di diabete, la malattia legata proprio all’incremento della glicemia, cioè del glucosio che circola nel sangue.
Un aumento dei valori (iperglicemia) può essere sintomo di
diabete mellito, pancreatite, insufficienza renale o potrebbe essere dovuto a
terapie a base di cortisonici o farmaci neurologici, analgesici, antipiretici,
antineoplastici. È aumentato inoltre nei tumori cerebrali, sindromi convulsive,
epatopatie croniche. Una sua diminuzione (ipoglicemia) potrebbe essere dovuta a farmaci, alcool, digiuno, Sindrome di Zollinger-Ellison, tumori maligni di grosse dimensioni, glicosuria renale. L'esame:Si effettua mediante prelievo del sangue a digiuno da almeno 12 ore.
è molto importante
che questo test venga fatto a riposo (oltre che a digiuno), in quanto uno stato
di stress può alterarne il risultato (falso positivo).
Risultati: Generalmente si considerano spie di un possibile
cambiamento nel normale equilibrio della glicemia valori inferiori a 70 e
superiori a 110 milligrammi per decilitro di sangue. Ma questo dato, da solo,
può tuttavia rendere opportuno eseguire controlli aggiuntivi. Infatti, in caso
di valori leggermente aumentati (120-140 mg/dl), è consigliabile eseguire una
curva di carico del glucosio(OGTT), cioè il controllo della glicemia dopo che
sono stati somministrati quantitativi fissi di glucosio in vena, per valutare la
risposta dell'organismo alla sollecitazione, in modo da evidenziare le persone
che presentano un'alterata tolleranza al Glucosio. Viene inoltre eseguito nelle
donne gravide al terzo mese, che presentano una glicemia a digiuno maggiore di
90-95 mg/dl. Infatti, diversi fattori, dallo stress psicofisico, al sovrappeso,
possono influenzare i valori dello zucchero in circolo. Per questo motivo, spesso il medico suggerisce questo test oppure la valutazione della glicemia durante l'arco di tutta la giornata, per stabilire se il pancreas, l'organo che produce l'insulina (ormone che ha il compito di "tamponare" il glucosio) risponde in modo adeguato agli stimoli. Organo ghiandolare in parte esocrino, in parte endocrino. La funzione esocrina consiste nella produzione di enzimi digestivi da immettere nel tubo digerente. La parte endocrina produce insulina e glucagone, riversati poi nel circolo sanguigno. Insulina: ormone proteico secreto dalle cellule beta delle isole di Langerhans del pancreas, che ha funzione anabolica nel metabolismo dei carboidrati, delle proteine e dei lipidi. Glucagone:ormone polipeptidico prodotto dalle cellule alfa delle isole di Langerhans del pancreas. Regola il metabolismo glicidico con azione iperglicemizzante, in antagonismo con l’ insulina. Il principale stimolo alla sua secrezione in circolo è costituito dall’ ipoglicemia: in tale situazione il glucagone agisce attivando vie metaboliche che fanno aumentare la concentrazione di glucosio nel sangue.
Poiché la produzione di energia mitocondriale rappresenta la vasta maggioranza di produzione di energia totale, la funzione mitocondriale è una funzione necessaria ed essenziale della regolazione del tasso metabolico basale. Cioè l'ormone tiroideo diminuito o la disfunzione mitocondriale può abbassare il tasso metabolico basale ed indurre i sintomi di ipotiroidismo (mani e piedi freddi, sensibilità a tempo freddo, depressione psicologica, difficoltà conoscitive, pelle asciutta, cuoio capelluto scaly, capelli fragili, problemi mestruali, costipazione, produzione diminuita di HCl dello stomaco, ecc.). l'insufficienza mitocondriale Non-tiroide-relativa ha potuto rappresentare facilmente l'alta incidenza dei sintomi ipotiroidei in individui con i livelli altrimenti-normali dell'ormone tiroideo. Forse una quantità significativa di ipotiroidismo infraclinico è insufficienza realmente mitocondriale. Senza riguardo a che cosa è denominata, la produzione di energia mitocondriale diminuita riduce la capienza della cellula alla funzione. Secondo le popolazioni delle cellule influenzate, questo può fare diminuire la temperatura corporea, abbassa la funzione immune, altera lo sviluppo, fa diminuire la riparazione del DNA, altera l'udienza, indebolisce la sintesi dei muscoli, dello steroide di diminuzione e del neurotrasmettitore ed abbassa i potenziali elettrici del sistema nervoso. Questi sono tutti i fattori che sono associati con entrambi i malattie ed ipotiroidismo mitocondriali. L'equilibrio fra il consumo di energia (calorie consumate) ed il dispendio energetico (calorie bruciate) è un fattore significativo che influenza il peso corporeo e la composizione. L'adeguatezza della funzione mitocondriale è essenziale ad effettuare un alto tasso metabolico basale e una massa magra del corpo. La produzione di energia mitocondriale dipende dai combustibili del grasso e del carboidrato. Carboidrati ( cioè. , gli zuccheri) sono il combustibile primario a causa della relativa disponibilità rapida. Grassi ( cioè. , i trigliceridi) sono il combustibile (di sostegno) secondario a causa della relativa idoneità ad immagazzinaggio ed è densità su calorica. Il grammo per il grammo, grasso contiene più di due volte l'energia del carboidrato. Il grasso è mobilitato quando il carboidrato è insufficiente per soddisfare le esigenze del corpo. I trigliceridi sono eliminati da immagazzinaggio e sono trasportati attraverso la circolazione sanguigna alle cellule in cui sono analizzati negli acidi grassi e nel glicerolo. Gli acidi grassi allora sono trasportati nei mitocondri da carnitina, in cui sono tagliati nelle piccole parti tramite un'beta-ossidazione denominata trattata. Queste parti (acetato) sono inserite nel ciclo dell'acido citrico per generare il trifosfato di adenosina e nel NADH per rifornire la catena di combustibile di trasporto dell'elettrone. L'ottimizzazione della funzione mitocondriale per migliorare la produzione di energia può dipendere dall'utilizzazione sia del carboidrato che dal grasso-bruciare le vie. Sappiamo che dipende dalle sostanze nutrienti critiche che sostengono la funzione mitocondriale. Nell'ultima edizione, abbiamo descritto i ruoli che 1) i giochi ALC e (della carnitina) in acido grasso trasportano nei mitocondri, 2) coenzima Q giochiamo nella catena di trasporto dell'elettrone, 3) giochi acidi lipoic nel ciclo dell'acido citrico, 4) NADH e gioco FADH2 nella coppia del ciclo dell'acido citrico alla catena di trasporto dell'elettrone e 5) le vitamine B-complesse giocano come coenzimi in molti di questi processi. Ma ci sono inoltre requisiti di biochemical/nutritional della produzione dell'ormone tiroideo che deve essere considerata pure. Le mancanze di queste sostanze nutrienti possono direttamente alterare la produzione dell'ormone tiroideo ed interferire quindi indirettamente con la funzione mitocondriale.
Il gene nucleare POLG codifica la subunità catalitica della DNA polimerasi gamma
mitocondriale. Nell'uomo tale gene presenta nel
primo esone una ripetizione trinucleotidica (CAG)5-13. L'allele più frequente in
popolazioni etniche diverse ha dieci triplette (frequenza =0.88) ed è assente
nel 1% degli individui. Le alterazioni della POLG possono contribuire
all'accumulo di anomalie nel DNA mitocondriale con
conseguenze sul metabolismo energetico.
Morbo di Parkinson e alterazioni della catena respiratoria mitocondriale. Negli ultimi anni l’attenzione dei ricercatori si è focalizzata sul possibile ruolo esercitato da fattori tossici esogeni o endogeni in alcune delle principali affezioni degenerative del sistema nervoso centrale. L’ipotesi eziopatogenica, che individua nei radicali liberi (ossidi e perossidi) i probabili responsabili del danno cellulare nel morbo di Parkinson, ha trovato nuovo vigore con la scoperta dell’azione lesiva del metabolita attivo della MPTP, MPP+ sui neuroni della Substantia Nigra, attraverso un blocco funzionale nella catena respiratoria mitocondriale. Nello studio, cui daremo inizio nel corso del 1999, noi useremo come substrato le piastrine, organelli ricchi di mitocondri e di facile prelievo anche durante fasi diverse della malattia parkinsoniana. I nostri dati preliminari hanno verificato la capacità delle piastrine di incorporare e metabolizzare L-Dopa. Nel modello piastrinico, quindi, verificheremo il possibile danno mitocondriale esercitato da cataboliti tossici della L-Dopa e della perossidazione lipidica. Al fine di identificare un possibile fattore tipo MPP+ verranno allestite colture cellulari, che saranno cimentate con omogenato proveniente da piastrine di pazienti con morbo di Parkinson che abbiano mostrato alterazioni della catena respiratoria mitocondriale.
SCOPERTO UN NUOVO GENE PER LA MALATTIA DI PARKINSON (http://www.telethon.it/comunicazione/)
Presso i laboratori dell’Istituto CSS Mendel di Roma, diretto dal Prof.
Bruno Dallapiccola, il gruppo di ricerca coordinato dalla dott. Enza
Maria Valente ha scoperto un nuovo gene responsabile di una forma di
malattia di
Parkinson ad esordio giovanile e trasmissione
recessiva (PARK6). La scoperta, frutto di un progetto finanziato da
Telethon e condotto in collaborazione con ricercatori inglesi e tedeschi, è
stata pubblicata sulla prestigiosa rivista “Science” .
Valutazione del metabolismo energetico in piastrine e colture di fibroblasti di soggetti affetti da sclerosi laterale amiotrofica familiare (SLA) e sporadica. Studi recenti attribuiscono assumere ai mitocondri un ruolo importante nel complicato processo degenerativo del motoneurone, non solo nelle forme familiari dell’ALS con mutazioni puntiformi sul gene delle SOD1, ma anche nelle altre forme familiari e in quelle sporadiche. Risulta inoltre evidente dalla letteratura uno stretto rapporto tra alterazioni dell’attività degli enzimi detossificanti da radicali liberi (come la SOD1) e il metabolismo energetico mitocondriale. Con lo studio che progettiamo di iniziare nel 1999 ci ripromettiamo di usare metodiche di coltura cellulare, tecniche istochimiche e biochimiche, per a) verificare mediante lo studio del metabolismo piastrinico un eventuale alterazione del metabolismo energetico mitocondriale in pazienti affetti da ALS sia familiare che sporadica; b) studiare l’effetto diretto del siero e del liquido cerebrospinale sul metabolismo energetico (prima e dopo esposizione ad agenti farmacologici o tossici) su piastrine e su colture di fibroblasti ricavati da pazienti con ALS; c) studiare l’eventuale effetto diretto di piastrine (o di un loro componente) ottenute da pazienti con ALS su colture di fibroblasti pure ricavati da pazienti con ALS.
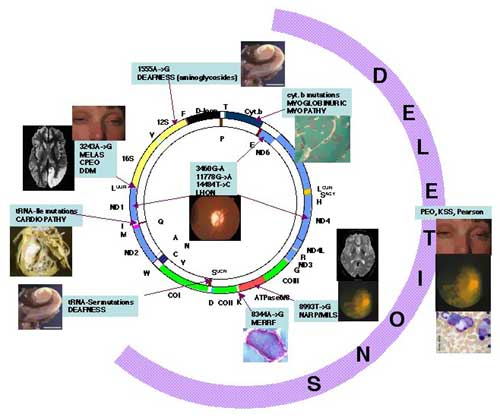
Mappa delle patologie del mtDNA. mutazioni corrispondenti. umeri e lettere si riferiscono alla posizione e al tipo di mutazione. Vedi anche mitomap.
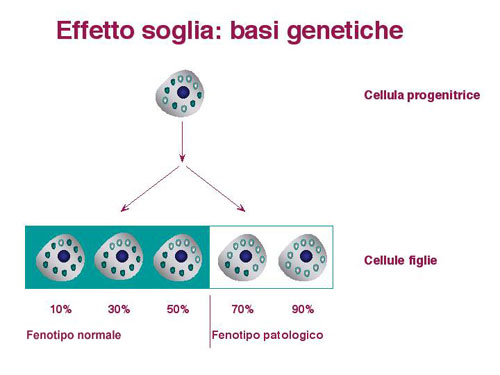
Esempio di "effetto soglia" in una linea cellulare. Gli organelli "pieni" rappresentano mitocondri contenenti mtDNA normale, quelli "vuoti" rappresentano mitocondri contenenti mtDNA patologico
|
|
|
Ultimo aggiornamento: 05-08-07