|
Come si
fa a diagnosticare una malattia mitocondriale?
L’intolleranza allo sforzo con accumulo di acido lattico a riposo.
La
presenza di alterazioni mitocondriali rilevabili dall’analisi della biopsia
muscolare.
La conferma
definitiva della diagnosi si ha però con il riscontro di un preciso difetto
biochimico degli enzimi della catena respiratoria o di una mutazione sul DNA
mitocondriale
Quali esami genetici
sono utili nelle malattie mitocondriali?
È consigliabile eseguire l’analisi delle mutazioni puntiformi del DNA
mitocondriale, tramite un semplice prelievo di sangue, in quelle famiglie dove
sia stato individuato almeno un caso di encefalopatia mitocondriale.
Poiché, anche all'interno della stessa famiglia, la variabilità dei sintomi può
essere elevata, la previsione sul decorso della malattia nei portatori di basse
percentuali di mutazione è difficilmente formulabile.
La diagnosi prenatale è spesso possibile, ma anche in questo caso la previsione
sul decorso della malattia non è sempre facilmente formulabile.
Non tutte le malattie mitocondriali
dipendono da alterazione del DNA dei mitocondri. Molte sono dovute ad
alterazioni in geni nucleari e si trasmettono come malattie monogeniche
Considerazioni Diagnostiche
La
diagnosi di malattia mitocondriale si basa sullo studio multidisciplinare del
paziente e dei suoi familiari, dal punto di vista clinico, morfologico,
biochimico e genetico-molecolare.
L'esame clinico accurato è fondamentale nell'indirizzo di tutta la successiva
procedura diagnostica. La presenza di miopatia progressiva e/o di
PEO,
spesso accompagnate da segni "centrali" coinvolgenti diversi sistemi
neuronali, è abbastanza tipica, ma spesso è
proprio l'associazione inusuale di segni clinici a carico di sistemi
ed organi embriologicamente e funzionalmente non correlati, in sindromi
cliniche complesse e di difficile inquadramento nosologico, a far
insorgere il sospetto di una patologia
mitocondriale. Il sospetto si rafforza se la sindrome è ereditaria, e vi è
evidenza di trasmissione materna.
Accanto alla patologia a carico del SNC (ad es. la sindrome di Leigh) e
del muscolo scheletrico (ad es. le
gravi miopatie infantili precoci da deficit isolato di COX), vi sono spesso
quadri atipici, con coinvolgimento prevalente od esclusivo di altri organi.
Ad esempio sono stati descritti quadri di grave
insufficienza epatica connatale
o infantile precoce, associata a steatosi e cirrosi micronodulare. Per quanto
riguarda il sistema cardiovascolare, casi di morte improvvisa, o
l'insorgenza di blocchi di branca,
o il "deficit di pompa" causato dalla presenza di una cardiomiopatia
progressiva non sono rari in pazienti "mitocondriali". Ciò non
è sorprendente dal momento che il cuore è un tessuto che consuma molta energia,
fornita in massima parte dal
metabolismo ossidativo aerobio. La biopsia endomiocardica si è dimostrata utile
in casi selezionati, per evidenziare alterazioni morfologiche,
biochimiche e molecolari. In campo
nefrologico, la nefropatia tubulo-interstiziale, ma soprattutto la
tubulopatia prossimale di DeToni-Fanconi sono le manifestazioni più
frequentemente osservabili in
patologia mitocondriale pediatrica. Nei pazienti tubulopatici può talora mancare l'acidosi lattica ma spesso un elevato acido lattico
urinario, così come valori anomali
degli intermedi del ciclo di Krebs, suggeriscono difetti della catena
respiratoria. Infine, il diabete mellito ad esordio precoce, ma anche
l'ipoparatiroidismo, l'ipotiroidismo, l'ipogonadismo ipogonadotropo, e il
deficit di ACTH, sono stati
segnalati quali complicazioni endocrinopatiche delle mitocondriopatie
pediatriche. Dal punto
di vista laboratoristico, il dato di più frequente riscontro è la presenza di elevati livelli di acido lattico nel sangue e, spesso, anche
nel liquor cefalo-rachidiano (LCR).
Particolarmente indicativo di alterazioni degli enzimi della catena respiratoria
è il contemporaneo rialzo di lattato, piruvato ed alanina, con elevato
rapporto lattato/piruvato. Al contrario, l'acidosi lattica associata
ad un basso indice lattato/piruvato
suggerisce la presenza di difetti della piruvato deidrogenasi. La
acidosi lattica è un dato di quasi costante riscontro nel bambino
"mitocondriale", mentre nell'adulto può essere assente. Data la difficoltà
della diagnosi clinica nel paziente
pediatrico, le misurazioni del lattato ematico e liquorale e degli altri
"punti redox" (acido
piruvico, beta- idrossibutirrato e acetoacetato) se eseguiti correttamente
da un laboratorio esperto, sono esami poco costosi e rapidi, che
risultano estremamente utili per l'inquadramento
diagnostico. Nei casi dubbi, può essere utile la
valutazione di curve del lattato "dinamiche" (durante e dopo un
esercizio muscolare standard
nell'adulto, o, nel bambino, durante il digiuno prolungato e in periodo
post-prandiale). Lo
studio morfologico della biopsia muscolare è fondamentale in quasi tutti i casi
di patologia mitocondriale,
pediatrici e adulti, anche se si è già sottolineato che talora i quadro morfologico muscolare è normale (ad es. nella LHON e
nella NARP). Il tessuto muscolare
è ideale per lacaratterizzazione biochimica dei difetti della catena
respiratoria, e per la rivelazione istochimica di deficit della COX.
Inoltre, la disponibilità di
tessuto muscolare bioptico è fondamentale in caso di fenotipi associati
amacroriarrangiamenti del mtDNA, in quanto tali alterazioni sono spesso
assenti in altri tipi cellulari (ad
es. leucociti ematici). Al contrario, nel sospetto di mutazioni puntiformi del
mtDNA, lo screening molecolare iniziale può essere eseguito anche su
DNA estratto da sangue periferico.
Le alterazioni neuro-radiologiche sono abbastanza tipiche. Si tratta
soprattutto di alterazioni simmetriche delle strutture cerebrali profonde
(anomalie del tronco encefalico, dei gangli della base, della sostanza
bianca periventricolare) e del
cervelletto (atrofia del verme o atrofia globale). Lesioni
encefalomalaciche, specie nelle zone posteriori degli emisferi cerebrali,
suggeriscono la
possibilità di una sindrome "MELAS". La spettroscopia basa sulla RMN
del cervello o del muscolo è stata
utilizzata per valutare, in maniera non invasiva, l'attività
metabolica e lo stato energetico di questi organi. E' pertanto possibil
identificare aree cerebrali o
gruppi muscolari in cui vi sia una eccessiva produzione di acido lattico o
una ridotta produzione di ATP. Ciò consente il follow-up non invasivo
dei pazienti e la
valutazione strumentale dell'eventuale efficacia dell'approccio terapeutico.
Poco é noto a questo riguardo ma
lo sviluppo di sistemi di ricerca avanzati, compreso
l'utilizzo di modelli animali transgenici e i primi tentativi di
"terapia genica", ed un più "disciplinato"
utilizzo di farmaci energetizzanti (carnitina, CoQ10, etc) forniranno nel ampie
possibilità terapeutiche.
L’interesse
per queste curiose e indispensabili strutture cellulari è aumentato ancor più
dopo la dimostrazione recente che un gruppo eterogeneo di gravi e numerose
malattie (note come "malattie mitocondriali") è causato da mutazioni del DNA dei mitocondri, ma è tuttora poco noto
come queste mutazioni possano
provocare differenti quadri patologici. Lo studio, condotto da Rosario Rizzuto
insieme a Paolo Pinton e Tullio Pozzan, in
collaborazione con ricercatori americani dell’Università del
Massachussetts, utilizzando un
approccio tecnologico innovativo (ossia un microscopio a
fluorescenza ultrarapido che permette di acquisire 30 immagini in meno di
un secondo), ha ottenuto per la
prima volta un’immagine tridimensionale di questi organelli in cellule viventi. I risultati di questo lavoro
cambiano una nozione ormai consolidata in biologia: i mitocondri non sono
infatti piccoli organelli "a forma di sigaro" distinti e indipendenti, come si era creduto
finora, ma un intricato reticolo interconnesso, in rapido e continuo movimento
all’interno della cellula. Questo
nuovo concetto può servire a comprendere non solo processi importanti nella
vita di questi organelli, ma anche come si sviluppano le malattie dovute a mutazioni del DNA
mitocondriale. Inoltre è stato possibile dimostrare che i mitocondri sono a
strettissimo contatto con un’altra struttura della cellula, il reticolo
endoplasmatico, e grazie a questa vicinanza ricevono, al momento opportuno, il
segnale di attivazione. Quando una cellula viene
stimolata, infatti, il reticolo endoplasmatico rilascia ioni calcio nelle
immediate vicinanze dei mitocondri,
che li captano prontamente e vengono così attivati.
Questo raffinato meccanismo di segnalazione e il ruolo fondamentale degli
ioni calcio nel controllo della funzione mitocondriale aprono l’affascinante
prospettiva di poterne "modulare" l’attività grazie allo
sviluppo di nuovi farmaci che
agiscano sul trasporto del calcio.
Per ulteriori informazioni rivolgersi al Dott. Rosario Rizzuto - Tel.
049/8276065 E’ disponibile
a richiesta copia dell’articolo pubblicato su Science
La visita neurologica
a cura della Redazione di DM
Patologie neuromuscolari diverse possono presentare talvolta sintomi e
segni simili. Una diagnosi precisa non può pertanto prescindere da un'accurata
ricostruzione del decorso della malattia e da un attento esame clinico, in altre
parole da un'accurata visita del paziente.
Importanti elementi per orientare la diagnosi sono l'età e il sesso, le modalità
di esordio del disturbo e l'andamento nel corso del tempo. Inoltre, quando si
sospetta una malattia ereditaria, è fondamentale indagare anche sugli altri
componenti della famiglia: possono infatti essere svelati eventuali ascendenti
che presentavano segni di una patologia miopatica (disturbi nella marcia,
difetti scheletrici o muscolari, ritardi psicomotori). Se i parenti risultano
clinicamente normali, sono opportuni studi del DNA, degli enzimi muscolari, i
dosaggi ormonali, l'elettromiografia o la biopsia muscolare: esami, questi, che
potrebbero rivelare anomalie non manifeste.
Prima di procedere all'ispezione diretta del paziente, si ritiene opportuno
porre allo stesso una serie di domande utili a evidenziare eventuali sintomi
magari non notati come tali: ipotrofie muscolari (scarso sviluppo di un
muscolo in un particolare settore del corpo), sensazione di tremori o
fascicolazioni (la "carne che balla"), dolori in varie sedi, urine scure
dopo sforzo, variazioni nella forza muscolare nell'arco della giornata o nel
caso di particolari attività.
E' anche utile un'indagine sulle abitudini di vita e sul tipo di attività svolte
dal paziente: questo infatti permette di valutare l'entità del disturbo e di
ricercare eventuali cause tossiche o traumatiche all'origine della patologia.
L'ispezione del paziente
Il primo elemento che viene osservato con attenzione in una visita ad un
paziente neuromuscolare è la deambulazione (la marcia) normale, poi
quella sulle punte e sui talloni.
La marcia anserina (a mo' di papera), associata a iperlordosi
(spostamento in avanti della colonna vertebrale all'altezza del bacino), rivela
una carenza di forza prossimale agli arti inferiori, cioè vicina all'asse
mediano del corpo (ad esempio dei muscoli delle cosce), tipica delle distrofie
muscolari. Di contro, una marcia steppante - cioè con un allungamento
accentuato della gamba - è segno di un deficit di forza distale, ovvero
agli arti inferiori (distanti dall'asse mediano del corpo: polpacci, piedi,
avambracci), come si osserva nelle polinevriti.
Si procede poi all'ispezione esterna del paziente svestito, prima in piedi, poi
disteso, prono e supino. Viene posta attenzione ad eventuali alterazioni della
pelle, deformità ossee o articolari (scoliosi, piede cavo ecc.), presenza di
asimmetrie delle masse muscolari, ovvero muscoli omologhi degli arti di diverse
dimensioni. Generalmente, in questo caso, si procede a misurazioni del diametro
dell'arto, tramite un metro, in sedi che siano alla stessa distanza da
determinati punti di riferimento ossei: ad esempio, per l'arto inferiore, la
rotula; in tal modo è possibile quantificare l'entità del fenomeno e
controllarne l'andamento nelle visite successive.
Un fenomeno ricorrente in caso di particolari malattie neuromuscolari può essere
l'atrofia del muscolo (muscolo non sviluppato), talvolta associata ad
astenia, cioè a debolezza muscolare. Non sempre comunque l'atrofia si
accompagna a debolezza: ad esempio, in alcuni processi di origine neurologica,
un'atrofia evidente può coesistere con una conservazione relativamente buona
della forza, mentre in altre patologie - come la miastenia e alcune miopatie
endocrine - si manifesta il processo inverso, cioè una forte astenia non
accompagnata da processi di atrofizzazione del muscolo.
Altri segni sono poi:
- le ipertrofie e le pseudoipertrofie muscolari, cioè uno sviluppo
abnorme, localizzato o generalizzato, della dimensione del muscolo.
Nel primo caso vi è un'associazione del segno clinico con debolezza muscolare.
Si parla invece di pseudoipertrofia quando la perdita di fibre muscolari
- che avviene solitamente nei processi distrofici - viene compensata da
proliferazione di materiale fibroso, di consistenza pastosa: è il caso della
pseudoipertrofia dei polpacci che si osserva nelle distrofie di Duchenne e
Becker, nelle atrofie spinali tipo Kugelberg-Welander e nelle donne portatrici
di distrofie tipo Duchenne;
- l'ipotrofia, cioè lo scarso sviluppo di gruppi muscolari. La sua
localizzazione in una sede particolare (cingolo scapolare, bicipite, tricipite,
muscoli della mano, muscoli distali degli arti inferiori) orienta di volta in
volta sulla probabile malattia di base;
- le retrazioni tendinee, cioè le riduzioni di lunghezza dei legamenti
che uniscono i muscoli alle ossa. Queste possono essere causate dal processo di
atrofizzazione (diminuzione di volume) del muscolo. Le conseguenze sono il
blocco delle articolazioni, in particolare delle caviglie, delle ginocchia, dei
gomiti e dei polsi. In pratica, i tendini si ritraggono limitando o impedendo
l'estensione completa della gamba. Tale fenomeno è frequente nei processi
distrofici e la sua precoce individuazione è importante per ritardarne
l'evoluzione con adeguati trattamenti fisioterapici;
- le facies, cioè le conformazioni del volto, determinate dai processi di
interessamento dei muscoli facciali. Esse possono comportare ptosi (abbassamento
delle palpebre), strabismo, palato ogivale, viso allungato;
- le fascicolazioni, cioè guizzi brevi (e per questo difficilmente
percepibili se non si presta una particolare attenzione) e aritmici, visibili
sotto la pelle che ricopre il muscolo in varie sedi del corpo. Esse possono
anche essere scatenate dalla percussione del muscolo, con il già menzionato
fenomeno della "carne che balla".
Il tono e la forza muscolare
Fin dalla fase neonatale si possono riscontrare segni di malattie neuromuscolari,
dovute a numerose affezioni ad esordio precoce. Uno di questi è la cosiddetta
ipotonia del lattante, vale a dire una debolezza muscolare che impedisce al
neonato di alzarsi. Il bambino ipotonico tende a rimanere disteso, a braccia e
gambe allargate (posizione "a rana"), e quando viene fatto muovere, gli arti e
le articolazioni sembrano allentati. Se il bambino inoltre viene sollevato per
le braccia in posizione seduta, il capo tende a cadere all'indietro, mentre, se
viene sostenuto per il ventre, la testa e gli arti pendono mollemente. Utili
indicazioni diagnostiche possono fornire anche l'intensità del pianto e della
respirazione.
Nei bambini più grandi e negli adulti il tono muscolare viene esaminato facendo
fare dei movimenti passivi alle estremità e valutando la resistenza che si
incontra. Per un test attendibile è necessaria, in questo caso, una buona
collaborazione da parte del soggetto.
In pazienti neuromuscolari l'esame della forza (eseguito opponendo una
resistenza al movimento compiuto dal paziente e saggiando segmento per segmento
i muscoli specifici o facendo compiere alla persona particolari "manovre") può
rivelare un'astenia (debolezza) localizzata in diversi punti:
- astenia generalizzata,
che coinvolge tutti i segmenti muscolari. E' propria della miastenia, delle
miopatie metaboliche e delle miopatie congenite;
- astenia prossimale,
che interessa i muscoli delle cosce, del cingolo pelvico (il bacino) o di quello
scapolare (le spalle), frequente nei processi miopatici e in alcune forme di
amiotrofia.
Nel caso di deficit di forza nei muscoli delle cosce o del bacino, i sintomi
saranno l'affaticabilità nella marcia, la facilità alle cadute, la difficoltà
nel sollevarsi da accovacciati o seduti e nel salire le scale senza appoggiarsi
a sostegni. La debolezza del cingolo scapolare può manifestarsi poi nella
difficoltà a pettinarsi o a stendere la biancheria, mentre l'esame clinico
rivela la presenza delle scapole alate, cioè sporgenti in modo rilevante;
- astenia distale, che colpisce i muscoli dell'avambraccio, del
polpaccio, della mano o del piede, ed è segno di patologie neurogene, salvo
alcune eccezioni, quali la miotonia di Steinert e alcune forme di miopatia dette
appunto distali.
Nel caso di un interessamento dei muscoli delle mani, il paziente avrà
difficoltà a compiere movimenti fini, come scrivere o adoperare le posate.
Quando invece sono coinvolti i muscoli degli arti inferiori, tenderà a
inciampare facilmente;
- astenia dei muscoli del tronco,
per la quale il paziente ha difficoltà a passare dalla posizione supina a quella
seduta senza fissarsi con le mani a qualche punto di appoggio (ad esempio il
bordo del lettino).
L'astenia può inoltre colpire i muscoli del collo, con la
difficoltà nel sollevare il capo e la tendenza a tenerlo lievemente flesso in
avanti, degli occhi, con diplopia (vista doppia) e caduta delle palpebre
(ptosi), del volto, con difficoltà nella masticazione, della
deglutizione e della fonazione, con modificazioni nel timbro della voce.
Mialgie e crampi
Tra i disturbi soggettivi lamentati da un paziente neuromuscolare, vanno
segnalate le mialgie (dolori muscolari), che si possono manifestare a
riposo o dopo sforzo (in questo caso si parla di crampi). Utile è anche
la palpazione delle masse muscolari per evidenziare eventuali mialgie dopo la
compressione delle stesse.
Dolori muscolari durante e dopo uno sforzo, che scompaiono con il riposo,
possono invece manifestarsi in patologie caratterizzate da carenza di enzimi
glicolitici muscolari. E' comunque sempre necessaria un'attenta valutazione da
parte del medico, in quanto le mialgie possono manifestarsi anche a seguito di
disturbi di origine psichica - come nevrosi o sindromi depressive - che non
hanno nulla a che vedere con patologie neuromuscolari.
Esame obiettivo generale
Un esame obiettivo generale dovrà ricercare la presenza di eventuali affezioni
cardiache, respiratorie, scheletriche, endocrine, metaboliche, infiammatorie,
neoplastiche, che si possono associare alle malattie neuromuscolari.
Va comunque segnalato che non sempre è sufficiente un esame clinico per
esprimere una diagnosi di malattia neuromuscolare. Talvolta, infatti, alcune
alterazioni enzimatiche, elettromiografiche o metaboliche sono presenti a
livello cellulare o molecolare anche di fronte ad un quadro clinico negativo.
Nel caso, ad esempio, di alcune donne portatrici della distrofia di Duchenne o
di Becker, solo un'accurata indagine genetica e l'analisi del DNA possono
svelare un'anomalia, poiché per il resto esse appaiono perfettamente sane.
Per questo, quindi, è necessario svolgere specifiche analisi strumentali quando
l'esame clinico obiettivo fa sospettare la presenza di una patologia
neuromuscolare.
Esami del sangue
CPK
La CPK (creatinfosfochinasi) - detta anche CK (creatinchinasi) - è un enzima
presente in modo caratteristico ed elevato nel muscolo e nel miocardio (la parte
muscolare del cuore). Si tratta di una proteina che ha la funzione di
trasformare in modo reversibile l'ATP (l'adenosin-trifosfato, sostanza che serve
al trasporto di energia) e la creatina (aminoacido presente nel tessuto
muscolare) in fosfocreatina, composto donatore di energia, essenziale per
la normale attività muscolare.
Sia la CPK muscolare che quella cardiaca sono composte da due sub-unità, che
possono essere di tipo M o di tipo B: nel muscolo si trova la CPK con doppia
sub-unità M (forma MM), mentre nel cuore si trova sia la forma MM che una forma
MB (sub-unità M associata a sub-unità B).
L'aumento della CPK nel siero (liquido del sangue che si separa da questo in
seguito alla coagulazione) può essere causato da alterazioni muscolari o anche
cardiache. Tra queste ultime va segnalato in particolare l'infarto del
miocardio, in cui la CPK si caratterizza per un aumento di quattro-cinque volte
il normale, nei due giorni successivi all'evento patologico, e per una
componente di forma MB particolarmente evidente.
Generalmente, tuttavia, specie nel bambino e nel giovane, la CPK aumenta in
seguito ad alterazioni del tessuto muscolo-scheletrico. Un'alterazione lieve e
transitoria è spesso dovuta a cause banali, quali un'iniezione intramuscolare,
uno sforzo prolungato o un trauma muscolare, ma un aumento persistente ed
elevato (spesso oltre dieci volte il normale) è caratteristico proprio delle
distrofie muscolari, con alterazioni della membrana delle fibre, in particolare
delle forme X-linked (distrofia di Duchenne, di Becker) e di altre forme, quali
la distrofia da deficit di adalina e la distrofia congenita da deficit di
merosina.
Nelle distrofie X-linked, l'analisi della CPK nel siero dopo sforzo muscolare
viene utilizzata per la diagnosi dello stato di portatrice, anche se bisogna
ricordare che a questo scopo l'analisi del DNA ha una capacità diagnostica molto
superiore. Da segnalare poi che incrementi dei dati di CPK si possono osservare
anche nella polimiosite e nell'ipotiroidismo. I valori più elevati si registrano
in seguito a rabdomiolisi acuta - ovvero a necrosi massiccia delle fibre
muscolari - fino ad arrivare anche a cento-trecento volte oltre il normale; tale
evento si configura sul piano clinico come crisi mioglobinurica (vedi oltre).
Normalmente, quindi, un aumento della CPK sierica è un segno di malattia
muscolare. E tuttavia va ricordato che alcune miopatie si possono generalmente
presentare con valori lievemente alterati o addirittura normali: tra questi, la
distrofia miotonica, la distrofia facio-scapolo-omerale e alcune forme di
miopatia congenita.
Nelle atrofie muscolari spinali e nelle polineuropatie, la CPK è di regola
normale, ma può presentarsi aumentata nelle forme croniche.
Nella distrofia di Duchenne, infine, la CPK sierica tende a decrescere con
l'età, anche se, fino a quando il paziente è ancora deambulante, si possono
osservare variazioni in aumento o in decremento che si collegano non al decorso
della malattia ma a fattori casuali, quale ad esempio la quantità di esercizio
muscolare eseguito il giorno prima del prelievo.
Latticodeidrogenasi (LDH) e altri enzimi
Nel siero di pazienti con malattie muscolari si può anche riscontrare l'aumento
del valore di altri enzimi presenti nelle fibre muscolari, e in particolare
della latticodeidrogenasi (LDH), dell'aldolasi, della piruvicochinasi (PK) e
dell'aspartatoaminotransferasi (AST).
Nelle miopatie in cui si rileva un aumento della CPK, tali enzimi si possono
presentare anch'essi alterati, ma di solito in modo meno evidente, cosicché la
loro valutazione riveste meno importanza nella diagnostica della patologia
muscolare. Va segnalato tuttavia che in alcuni casi, come ad esempio in alcune
forme o fasi della polimiosite, specie infantile, si può osservare nel siero una
CPK normale, accanto all'alterazione isolata di altri enzimi quali la LDH.
L'elevazione degli enzimi AST e ALT (alaninaaminotransferasi) è considerata
espressione di malattia epatica, dato che il fegato ne è particolarmente ricco:
ed è per questo che quando, in caso di miopatia, aumentano nel sangue le
transaminasi, e in particolare la AST, il medico può essere erroneamente
indirizzato verso una malattia del fegato, se contemporaneamente non si procede
ad una valutazione della CPK.
Acido lattico plasmatico
Alcune malattie muscolari - in particolare le miopatie metaboliche - possono
causare un alterato valore dell'acido lattico plasmatico, valutato a riposo o
nell'ambito di particolari prove.
Nel tessuto muscolare, l'acido piruvico, quando non può entrare nei mitocondri
per essere ossidato, viene trasformato in acido lattico. Questo permette che la
glicolisi, a monte, continui a trasformare altro glucosio in acido piruvico e
così produrre legami energetici. Un aumento dell'acido lattico - o dell'insieme
di acido lattico e acido piruvico - in generale è indice di un'alterazione
dell'attività mitocondriale, quale si può avere per deficit della
piruvicodeidrogenasi, della piruvicodecarbossilasi, della citocromo-C-ossidasi o
di altri enzimi della catena respiratoria. Queste anomalie enzimatiche spesso si
manifestano come encefalomiopatie, cioè malattie a prevalente coinvolgimento
muscolare e cerebrale.
Nel sospetto di una patologia mitocondriale, se l'acido lattico basale è
normale, si può evidenziare un suo incremento patologico mediante la "Prova da
sforzo aerobico", test nel quale il paziente esegue un esercizio di venti
minuti alla cyclette e viene sottoposto poi a una serie di prelievi di sangue
seriali proprio per dosare l'acido lattico.
L'acido lattico plasmatico può anche essere valutato nella "Prova da sforzo
anaerobico", test in cui il paziente apre e chiude ripetutamente con
forza una mano per un minuto, mentre viene bloccata la circolazione arteriosa
del braccio (e quindi l'apporto di ossigeno) mediante un bracciale.
Una successione di prelievi allo stesso arto viene eseguita prima e dopo il
test: se essi indicano una mancata produzione di acido lattico, sono segnale di
alterazione della glicolisi o della glicogenolisi. Ne sono esempi le miopatie da
deficit degli enzimi fosfofruttochinasi e miofosforilasi.
Mioglobinemia
Il trasporto di ossigeno all'interno delle fibre muscolari è regolato da un'emo-proteina
denominata mioglobina. In generale, le malattie muscolari che determinano
aumento nel siero delle CPK comportano anche il concomitante aumento della
mioglobina: nella diagnostica delle malattie neuromuscolari, il dosaggio
plasmatico di questo costituente appare pertanto poco rilevante, se si
eccettuano i casi dubbi di mioglobinuria (vedi oltre).
Il dosaggio della mioglobinemia (concentrazione di mioglobina nel sangue) è più
utile nella diagnosi dell'infarto miocardico, perché questo evento determina un
aumento della mioglobina nel plasma più precocemente di quello della CPK.
Carnitinemia
Presso alcuni centri specializzati si può dosare nel sangue la carnitina, una
sostanza che viene in parte assunta col cibo e in parte sintetizzata
nell'organismo. Essa ha la funzione di trasportare gli acidi grassi a lunga
catena all'interno dei mitocondri per l'ossidazione. Per questo la mancanza di
carnitina determina accumulo di grassi nei tessuti.
Nella miopatia da deficit di carnitina, questa sostanza è deficitaria nel
muscolo ma presente nel sangue. Si osserva invece un livello molto basso di
carnitinemia (concentrazione di carnitina nel sangue) nella forma sistemica
della malattia da deficit di carnitina, patologia molto rara in cui oltre al
muscolo scheletrico sono interessati anche altri organi, quali cuore, reni,
cervello e fegato.
Si possono osservare riduzioni della carnitinemia in conseguenza di altre
malattie, come ad esempio nei disturbi del metabolismo del piruvato (altra
denominazione dell'acido piruvico salificato), o in forme di encefalomiopatia
mitocondriale. Più frequentemente, questa condizione di deficit secondario di
carnitina si può osservare in seguito ad emodialisi, malnutrizione, gravidanza o
trattamenti con particolari farmaci.
Anticorpi antirecettori dell'acetilcolina
E' quest'ultimo un esame utile nella diagnostica della miastenia grave, malattia
causata da autoanticorpi diretti contro i recettori dell'acetilcolina della
placca neuromuscolare (punto di contatto tra il nervo e la fibra muscolare).
L'identificazione di tali anticorpi nel siero indica la diagnosi di miastenia
grave.
Va tenuto presente che circa il 10-20% dei pazienti miastenici non sono positivi
a questo esame e pertanto la normalità dello stesso non può escludere la
presenza della malattia.
Altre indagini nel sangue
Oltre a quanto elencato, a livello ematico si possono eseguire anche altri esami
ai fini della diagnosi di una malattia neuromuscolare. Tra questi ricordiamo
l'analisi degli acidi organici, che va eseguita anche nelle urine, e che può
permettere l'identificazione di malattie metaboliche molto gravi.
Esami delle urine
Mioglobinuria
Il muscolo scheletrico è ricco di mioglobina, un'emoproteina simile
all'emoglobina del sangue, che è in grado di trasportare ossigeno dal capillare
al mitocondrio, dove viene utilizzato per l'ossidazione metabolica. La
mioglobinuria (comparsa di mioglobina nelle urine) è conseguente alla presenza
nel plasma di una mioglobinemia in valori superiori a quelli della soglia renale
per questo pigmento.
Tale evento patologico è possibile quando dal muscolo vengono rilasciate nel
sangue massicce quantità dei suoi costituenti, generalmente in seguito a necrosi
muscolare acuta diffusa (rabdomiolisi), come si osserva nelle crisi
mioglobinuriche ricorrenti (la mioglobinuria è la presenza di mioglobina nelle
urine), caratteristiche di alcune malattie metaboliche.
Tra queste ultime la più frequente è la forma genetica da deficit dell'enzima
detto carnitina-palmitil-transferasi. Ma crisi mioglobinuriche possono
verificarsi anche in pazienti con distrofia, ad esempio dopo sforzi muscolari
eccessivi, e possono rappresentare l'unico sintomo di una forma X-linked
(variante mite della distrofia di Becker). La mioglobinuria, infine, può anche
essere causata da schiacciamento di estese masse muscolari, come succede - per
citare un esempio semplice, ma particolarmente evidente - in seguito ai traumi
derivanti dal crollo di un edificio.
Nella crisi mioglobinurica conclamata, le urine presentano un caratteristico
color "coca-cola" (una volta si parlava di color "marsala"), mentre non cambiano
molto il colore se la quantità di mioglobina nelle urine è modesta. In ogni caso
esami immunologici o spettrofotometrici specifici possono indicare con
precisione la quantità di mioglobina presente nelle urine.
Nei laboratori meno attrezzati, la mioglobina viene evidenziata con test più
semplici che però non la distinguono dall'emoglobina del sangue. In questi casi
sarà solo il contemporaneo elevato aumento della CPK nel siero a indicare che si
è in presenza di mioglobinuria (mioglobina nelle urine) e non di emoglobinuria
(emoglobina nelle urine).
La diagnosi di crisi mioglobinurica è molto importante perché tale evento
morboso può essere gravissimo a causa di complicanze renali o cardiache. Le
forme più gravi si osservano per lo più in pazienti che presentano la cosiddetta
suscettibilità all'ipertermia maligna, collegata, quest'ultima, all'uso di
dialogenati o succinilcolina, sostanze usate per l'anestesia generale.
Queste stesse sostanze possono provocare gravi complicazioni anche in altri
pazienti miopatici, sia pure con minore probabilità. La presenza di una miopatia
deve portare quindi ad un'accurata valutazione anestesiologica, prima di
qualsiasi intervento chirurgico.
Altri accertamenti nelle urine
Nella diagnostica delle malattie muscolari, si possono eseguire anche altre
indagini di laboratorio a livello delle urine. In centri altamente
specializzati, si può ad esempio valutare il profilo urinario degli acidi
organici mediante metodiche diagnostiche molto sofisticate, come la
gascromatografia e la spettrofotometria di massa, esami espletabili anche a
livello del sangue.
Le analisi degli acidi organici consentono di diagnosticare le cosiddette
organicoacidurie, patologie rare, ma molto gravi, tra cui vanno segnalate le
malattie da alterazioni enzimatiche collegate al ciclo di Krebs (importante via
del metabolismo mitocondriale) - quale ad esempio la forma da accumulo di
acido metilmalonico - e quelle da deficit enzimatici della beta-ossidazione,
quale ad esempio la glutaricoaciduria di tipo II.
Biopsia:
-
La biopsia muscolare
permette di distinguere la natura della
miopatia, che può
essere, ad esempio, degenerativa, metabolica o infiammatoria.
-
La biopsia di nervo
periferico risulta indicata laddove la diagnosi clinica richieda una
caratterizzazione di alterazioni patologiche associate a vasculite, amiloidosi
e neuropatie ereditarie.
-
La biopsia di cute
si rivela utile nell’integrazione dello studio diagnostico di alcune
malattie neuromuscolari, quali ad esempio distrofia muscolare di
Emery-Dreifuss e distrofia neuroassonale.
-
Sul prelievo bioptico
possono essere effettuati studi istochimici, istoenzimatici,
immunoistochimici in microscopia ottica, ed ultrastrutturali in
microscopia elettronica.
La biopsia muscolare
di Marina Fanin
Come e perché
La biopsia muscolare è il prelievo di una limitata
porzione di muscolo (1 o 2 centimetri) dalla coscia o dal braccio, tramite una
piccola incisione in anestesia locale (simile a quella utilizzata dal dentista).
La ferita viene poi chiusa con qualche punto di sutura.
L'intervento per biopsia richiede circa mezz'ora di
tempo, ma solo di rado viene eseguito ambulatorialmente. Di solito, infatti, il
paziente che vi si sottopone viene ricoverato in ospedale per qualche giorno,
perché è necessario eseguire previamente una visita neurologica e specifici
esami del sangue, oltre ad un elettromiogramma, un elettrocardiogramma e una
radiografia del torace.
Eccettuati quei rari casi in cui il parere del
medico sia contrario, tutte le persone possono essere sottoposte a biopsia
muscolare senza correre rischi, compresi i bambini al di sotto di un anno di
vita.
La biopsia muscolare viene eseguita quando un
neurologo o un pediatra la ritengono necessaria. Infatti, si arriva a praticarla
quando la visita neurologica rileva dei sintomi sospetti che vengono confermati
dal risultato di altri esami mirati. Quelli che in genere il neurologo prescrive
sono l'elettromiogramma ed alcune analisi del sangue, che servono, ad esempio, a
conoscere il valore della creatin-kinasi (CK), enzima caratteristico
delle cellule muscolari.
In qualche caso, in cui un familiare soffra di una
malattia muscolare ereditaria, la biopsia può servire anche ad individuare altre
persone della stessa famiglia che - pur non soffrendo di quella stessa malattia
- la possono trasmettere
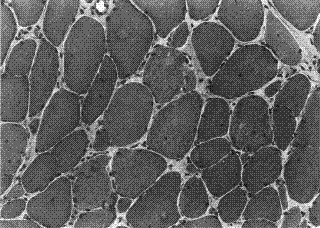
Sezione trasversale di biopsia muscolare, colorata con
ematossilina-eosina (duecento ingrandimenti). Alcune anomalie presenti
in questa biopsia sono costituite dalla coesistenza di fibre di dimensioni
maggiori della norma (fibre ipertrofiche) e di fibre di dimensioni
inferiori alla norma (fibre ipotrofiche o atrofiche), dalla presenza
di nuclei cellulari localizzati all'interno del citoplasma (nuclei
centrali) - anziché lungo la membrana cellulare - e dall'aumento del
tessuto connettivo interstiziale che separa le fibre muscolari tra di loro.
E' consigliabile sottoporsi all'intervento in un
centro specializzato in malattie neuromuscolari, dove vengano analizzate alcune
centinaia di biopsie per anno. Questo perché le procedure di indagine sono molto
sofisticate ed eseguibili solo nei centri più grandi e meglio attrezzati. Ciò
permette inoltre di condurre un ampio numero di esami sulla porzione di muscolo
prelevata. In qualche caso, infatti, le indagini più comuni non sono sufficienti
a far capire con esattezza di quale tipo di malattia si tratti, ed è perciò
necessario approfondire le analisi in modo ancora più specialistico.
Talora, per poter arrivare alla diagnosi corretta, è
necessario analizzare alcuni dei componenti del muscolo (proteine ed enzimi) che
risultano difettosi in determinate malattie, e queste ricerche si possono
eseguire solo sul prelievo effettuato tramite la biopsia.
I recenti progressi nel campo delle malattie
neuromuscolari ereditarie possono portare a chiedersi se sia sempre necessario
arrivare alla biopsia muscolare per ottenere una diagnosi. Si può dire
senz'altro che per diagnosticare la maggior parte delle distrofie muscolari - le
miopatie congenite (miopatia nemalinica, miopatia centronucleare,
miopatia central-core), le miopatie metaboliche, le miopatie
infiammatorie (miositi), le atrofie muscolari spinali e
la sclerosi laterale amiotrofica - tale strumento di indagine resti
insostituibile.
La diagnosi istologica
Appena prelevato, durante la biopsia, il muscolo
viene congelato immergendolo in azoto liquido (a una temperatura di -180 gradi).
Questa procedura di fissazione consente di:
a) ottenere delle "fette" (o "sezioni") di muscolo
estremamente sottili (dello spessore di millesimi di millimetro), adatte ad
essere successivamente colorate e osservate al microscopio;
b) utilizzare il pezzettino di muscolo per indagini
specialistiche (analisi biochimiche di enzimi muscolari, analisi sulle proteine
muscolari ecc.) quando, dopo le indagini preliminari, esse si rendano necessarie
per precisare la diagnosi;
c) conservare nel tempo il reperto rimanente, una
volta effettuate le analisi: infatti, se trattata e conservata correttamente in
particolari congelatori, una biopsia muscolare può essere utilizzata anche dopo
qualche decina di anni.
Come già accennato, la diagnosi istologica della
biopsia muscolare si ottiene tramite l'osservazione al microscopio di un numero
variabile di vetrini, sui quali vengono racchiuse e colorate alcune "fette" del
reperto. Ciascuno dei vetrini viene colorato in un modo diverso, per poter
evidenziare i vari componenti del muscolo.
Più ampio è il numero di colorazioni o di analisi
che vengono eseguite (analisi istochimiche ed
istoenzimatiche), più numerose sono le informazioni che si possono
ottenere.
Le analisi istochimiche più comuni (ematossilina-eosina,
tricromica, PAS, Oil Red O) sfruttano la proprietà chimica
delle soluzioni coloranti di legarsi in modo specifico ad uno dei componenti
delle cellule muscolari.
Con tali colorazioni si possono esaminare la forma e
la grandezza delle cellule muscolari, la posizione dei nuclei all'interno delle
cellule stesse, l'infiammazione del muscolo, l'aumento di materiale fibroso
non-muscolare (connettivo), la presenza di segni che indicano la distruzione
delle cellule (degenerazione o necrosi), la formazione di nuove cellule
(rigenerazione), l'aumento di particolari componenti (glicogeno, grassi).
Mentre nel muscolo normale le cellule sono di forma
e dimensione regolari, nel muscolo malato si possono osservare cellule dal
diametro più piccolo (atrofiche) o più grande (ipertrofiche) della
norma, cellule dalle forme schiacciate (angolate) anziché circolari,
nuclei localizzati all'interno della cellula anziché lungo il contorno
periferico (membrana).
Alcune particolari colorazioni istoenzimatiche (deidrogenasi
mitocondriali, citocromo ossidasi ecc.) permettono di verificare il corretto
funzionamento di alcuni componenti della cellula muscolare - chiamati
mitocondri - in cui avviene la produzione di energia utilizzata dalla
cellula stessa durante la contrazione del muscolo.
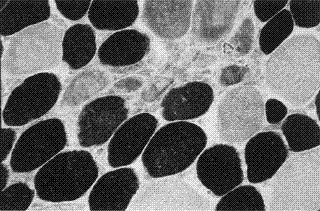 Sezione trasversale di biopsia muscolare, colorata con
l'enzima ATP-asi acida (duecento ingrandimenti). Si osservano fibre
muscolari che reagiscono positivamente e vengono identificate come fibre di
tipo 1 (fibre scure) e fibre di tipo 2 che non si colorano (fibre
chiare). Le fibre muscolari che rigenerano presentano con questa
reazione una colorazione di tipo intermedio.
Altre colorazioni istoenzimatiche (ATP-asi
miosiniche) consentono di evidenziare eventuali disturbi dell'innervazione del
muscolo che non si potrebbero osservare semplicemente con le colorazioni
istochimiche.
Dopo aver esaminato al microscopio tutte le
colorazioni istochimiche ed istoenzimatiche disponibili e dopo avere annotato le
eventuali anomalie osservate in ciascuna colorazione, un osservatore
sufficientemente esperto in malattie muscolari può formulare la diagnosi
istologica.
Va ricordato in conclusione che se queste sono le
indagini principali che si eseguono relativamente alla biopsia muscolare, in
qualche caso, però, può essere necessario approfondire li esami e procedere con
successive colorazioni immunoistochimiche, oppure con dosaggi enzimatici,
analisi di proteine, analisi sul DNA e altro ancora.
Tecniche neuroradiologiche
di Elisabetta Menegazzo
A fianco dei tradizionali esami di "routine"
(esame clinico, elettromiografia, biopsia), è opportuno segnalare anche
l'esistenza di tecniche neuroradiologiche per la diagnosi di patologie in cui si
sospetti un coinvolgimento del tessuto cerebrale.
Con il termine
tecniche neuroradiologiche si intendono in
particolare la risonanza magnetica nucleare (RMN) e la tomografia ad
emissione di positroni (PET).
RMN
In una radiografia tradizionale, le radiazioni
evidenziano più la struttura ossea e meno i tessuti. Per semplificare, si può
dire che la struttura ossea appare come la "parte chiara" dell'immagine, mentre
i tessuti appaiono "scuri".
Nel caso della RMN anche i tessuti non ossei
esaminati inviano "segnali" che vengono rilevati in presenza di un campo
magnetico e che danno luogo ad un'immagine.
Utilizzando opportunamente la RMN è possibile
ottenere un'immagine costituita da diverse gradazioni di grigio, che
corrispondono alle differenti densità del tessuto studiato.
L'esecuzione dell'esame in questione è semplice e
non invasiva. Il paziente viene posizionato orizzontalmente all'interno di una
macchina a forma di "tubo" e viene sottoposto a un campo magnetico. E' quindi
importante che egli non soffra di claustrofobia (paura dei luoghi chiusi), e che
non abbia in corpo schegge metalliche, protesi artificiali del cristallino,
pompe di infusione di insulina o pace-maker che possono portare ad effetti
dannosi, se sottoposti al campo magnetico.
PET
La PET è una metodica che si serve di un indicatore
(fluoro-desossi-glucosio) che è utilizzato dal tessuto cerebrale e che si
mescola nel sangue.
Si può valutare quindi come viene utilizzata tale
sostanza dalle cellule cerebrali.
Le patologie interessate
Essendo un esame non invasivo, la RMN è facilmente
eseguibile in età pediatrica per la diagnosi di malformazioni o alterazioni
cerebrali, che possono essere presenti nelle distrofie muscolari congenite.
E' anche utile nella diagnosi differenziale tra la
sclerosi laterale amiotrofica (malattia caratterizzata da progressivo
"dimagramento" dei muscoli, con mancanza di forza e sensazione di "carne che
balla"), la mielopatia da spondiloartrosi e la siringomielia
(caratterizzata da una "cavità" che si forma nel midollo spinale).
RMN e PET trovano applicazione anche nella diagnosi
della distrofia miotonica e delle encefalopatie mitocondriali.
La distrofia miotonica è una patologia
neuromuscolare in cui - oltre al tessuto muscolare - possono essere coinvolti
altri apparati, tra cui quello cerebrale. Tra le caratteristiche più importanti
di essa, vi è l'abnorme espansione di una sequenza di DNA situata nel cromosoma
19 (chiamata tripletta CTG).
In questo caso la RMN cerebrale può evidenziare
alterazioni della densità cerebrale e la PET alterazioni diffuse o localizzate
del metabolismo energetico (vale a dire dell'utilizzazione di sostanze
energetiche, quali il glucosio).
Riguardo poi alle encefalopatie mitocondriali,
esse sono sempre malattie neuromuscolari, dovute però ad un'alterazione del DNA
mitocondriale. Qui, all'interessamento del tessuto muscolare, si possono
associare disturbi di diverso tipo che coinvolgono anche il tessuto cerebrale.
Nelle encefalopatie mitocondriali, la RMN può
evidenziare la presenza nel cervello di aree con alterata circolazione sanguigna
(le cosiddette ischemie), mentre la PET consente di individuare aree in
cui l'utilizzazione di sostanze energetiche (metabolismo energetico) è
anomala.
L'elettromiografia clinica
a cura della
Redazione di DM
Il termine elettromiografia
(EMG) indica un particolare esame che amplifica, visualizza e registra
l'attività elettrica del muscolo volontario (muscolatura striata),
attività che è alla base del processo di contrazione del muscolo stesso, e la
risposta di un nervo a una stimolazione elettrica.
Esso viene praticato quando si
sospetta una patologia del muscolo (distrofia, miotonia), della giunzione
muscolo-nervo (miastenia), del nervo periferico, della radice del nervo o del
motoneurone.
Per chiarire con un esempio, l'EMG
registra quanta corrente passa attraverso un "filo elettrico" (il
motoneurone e il nervo periferico) e come funziona la "lampadina" che
deve accendere quel medesimo filo elettrico (il muscolo striato).
L'EMG consente quindi di
evidenziare la presenza di anomalie dei muscoli ("lampadina rotta") o di
patologie che provocano una ridotta o assente innervazione del muscolo ("filo
elettrico = cattivo conduttore" oppure "conduttore spezzato"): spesso, grazie a
questo tipo di indagine, è possibile localizzare la sede precisa di una lesione
nervosa. L'esame fornisce inoltre elementi obiettivi che permettono di
quantificare e di seguire l'evoluzione di un'affezione nervosa. Esso non
consente però di riconoscere la natura esatta del danno, nervoso o muscolare che
esso sia.
Il muscolo volontario
Per capirne un po' di più può
forse tornare utile qualche elementare nozione sulla struttura del muscolo e sui
suoi meccanismi di funzionamento.
Il muscolo volontario è formato
da gruppi di fibre muscolari disposte secondo un ordine regolare - le cosiddette
unità motorie - ciascuna delle quali innervata da un
motoneurone.
A sua volta, una fibra
muscolare è formata da unità ancora più piccole, le miofibrille,
costituite da filamenti microscopici, chiamati actina e miosina,
proteine che controllano la contrazione. Ogni fibra muscolare è fornita di una
terminazione nervosa che riceve gli impulsi che dal cervello vanno al midollo
spinale (1°
motoneurone) e dal midollo al muscolo (2°
motoneurone). L'impulso nervoso, simile ad una scarica elettrica,
stimola il muscolo liberando un neurotrasmettitore, una sostanza chimica
chiamata acetilcolina. Tutto questo dà inizio a una catena di eventi
chimici ed elettrici che provocano lo scivolamento dei filamenti di miosina su
quelli di actina, in modo molto simile ad una scala che si possa allungare o
accorciare. E' proprio tale movimento a determinare l'accorciamento
(contrazione) del muscolo.
La forza della contrazione del
muscolo dipende da due fattori: la frequenza delle pulsazioni delle singole
unità motorie (potenziale) e il numero delle unità motorie coinvolte nella
contrazione. In altri termini, più frequenti sono le pulsazioni e maggiore è il
numero delle unità motorie, più forte sarà la contrazione dell'intero muscolo.
La contrazione massima si avrà quando tutte le unità motorie saranno coinvolte
nel movimento e quando la pulsazione delle fibre sarà al livello massimo.
Le attività rilevate
Come detto all'inizio, l'esame
elettromiografico rileva l'attività elettrica del muscolo, vale a dire le
variazioni di potenziale delle unità motorie nel muscolo in attività, quando
esso passa da una fase di rilasciamento alla contrazione. Inoltre, esso permette
di stabilire - nel caso di una patologia che colpisce il sistema muscolare - se
il danno è di tipo neurogeno (atrofia neurogena,
provocata dalla lesione del
motoneurone, della radice o del nervo) o muscolare (atrofia
miogena, provocata dal danneggiamento di alcune fibre muscolari).
In un muscolo normale a riposo,
non c'è alcuna attività elettrica: il cervello (la cosiddetta "centrale
elettrica") non trasmette impulsi e non c'è pertanto alcun passaggio di corrente
attraverso i nervi (i cosiddetti "fili elettrici"). Nel corso del movimento
volontario graduato, si registra un'attività via via più complessa. Dapprima,
per un movimento lieve, si può ottenere l'attivazione di una sola unità motoria
con pulsazioni poco frequenti. Ciò permette di studiare le caratteristiche del
potenziale di unità motoria. A mano a mano che il movimento si va rinforzando,
si attivano nuove unità motorie (è il fenomeno definito sommazione spaziale)
e, nel contempo, ogni singola unità motoria aumenta la frequenza delle proprie
pulsazioni (sommazione temporale).
Un segnale di attività
spontanea in un muscolo a riposo non è normale: la sua presenza, infatti, può
indicare un'affezione acuta o cronica del nervo periferico, una rigenerazione
nervosa o una sindrome miotonica.
Solitamente un tracciato troppo
ricco di una debole ampiezza (polifasico), in un muscolo in contrazione,
fa pensare a una patologia muscolare. Al contrario, un tracciato povero e
accelerato, con dei potenziali talvolta di grande ampiezza, orienta verso
un'affezione nervosa.
L'esame di stimolo-rivelazione
misura il tempo che impiega la stimolazione elettrica a raggiungere il punto di
registrazione dello stimolo stesso. Stimolando più punti sullo stesso nervo si
può calcolare la velocità di conduzione nervosa (motrice o sensitiva):
eventuali anomalie sono segno di una neuropatia.
Le apparecchiature
La strumentazione utilizzata
per compiere l'elettromiografia comprende:
- gli elettrodi,
piccole "sonde" che vengono infisse nel muscolo da esplorare. Normalmente
l'elettrodo utilizzato è l'ago coassiale di Bronck, che ha al suo interno
uno o due fili (si parla, nel primo caso, di derivazione monofilare, nel
secondo di derivazione bifilare);
- gli amplificatori,
che amplificano appunto le differenze di potenziale raccolte dagli elettrodi;
- due oscillografi
catodici, sorta di "sismografi" nei quali viene immesso il segnale
amplificato e che consentono la scopia, cioè la visualizzazione immediata
delle variazioni di potenziale e la registrazione (fotografica o digitale) del
segnale;
- un altoparlante,
attraverso il quale esce una segnalazione acustica delle variazioni di
potenziale.
Come si esegue l'EMG
L'esame elettromiografico non è
standardizzato e la condotta di esso varia inizialmente a seconda del sospetto
diagnostico fondato sull'esame clinico e sui dati neurologici, oltre che -
successivamente - nel corso della stessa EMG, a seconda dei dati che si vanno
via via raccogliendo.
La posizione migliore per
l'esame è quella che consente al paziente il maggior rilassamento e al medico la
maggior libertà di azione possibili: ciò si ottiene con il paziente seduto o
semisdraiato su una poltrona a schienale regolabile, fornita di comodi braccioli
e sufficientemente lunga da consentire l'estensione completa degli arti
inferiori. Questa distensione condiziona la facilità e la rapidità dell'esame,
che solitamente dura fra i 30 e i 60 minuti.
Per l'esame di rivelazione
viene applicato un elettrodo nei muscoli che devono essere analizzati, scelti in
base all'esame clinico. Il paziente deve collaborare, di volta in volta
contraendo o rilassando il muscolo analizzato.
L'elettrodo è collegato a una
speciale macchina che registra i potenziali muscolari a riposo o durante la
contrazione e li visualizza attraverso gli oscillografi, che delineano un
tracciato. In un secondo tempo, lo studio della conduzione nervosa motoria o
sensitiva viene fatto stimolando il nervo attraverso dei deboli impulsi
elettrici e registrandone il segnale.
Si tratta certo di un esame
poco piacevole, ma facilmente tollerabile. Alcune persone sopportano con
difficoltà le stimolazioni elettriche, altri le punture per l'applicazione degli
elettrodi.
Piuttosto dolorose sono alcune
indagini come la stimolazione tetanica (forte contrattura
provocata artificialmente) del muscolo, talora utilizzata per rilevare una
miastenia; in altri casi si esegue a tal fine l'analisi della singola fibra (jitter)
che richiede un agoelettrodo particolare.
Diversi
elementi, che però non devono necessariamente coesistere, possono suggerire
o indirizzare la diagnosi. Tra quelli clinici vi sono l'intolleranza allo
sforzo (con un accumulo di acido lattico a riposo o in seguito a uno
sforzo), l'ereditarietà materna e la patologia multisistemica. Dal punto di
vista morfologico, la presenza di alterazioni mitocondriali rilevabili
dall'analisi della biopsia muscolare è indicativa della malattia. Un
indicatore biochimico è il deficit di uno o più enzimi mitocondriali, anche
questo riscontrabile dalla biopsia muscolare.
A rendere possibile la diagnosi definitiva è comunque il riscontro di una
mutazione del DNA mitocondriale. Mentre le delezioni devono essere accertate
sul DNA mitocondriale estratto dal muscolo, le mutazioni puntiformi sono
presenti in elevata percentuale in tutti gli organi, e sono quindi
facilmente identificabili in tutti i tessuti. In genere c'è una relazione
tra la percentuale di mutazione quantificata nel muscolo e la gravità del
fenotipo clinico.
Dal notiziario dell'AISLA n°3 2001
C'è da puntualizzare che la Malattia del Motoneurone (MND), non coincide
con la SLA, in quanto quest'ultima è compresa nel più ampio gruppo delle MND,
che annovera anche la Sclerosi Laterale Primaria (Primary Lateral
Sclerosis, PLS), L'Atrofia Muscolare Spinale Progressiva (Progressive
Spinal Muscular Atrophy, PSMA), le Atrofie Muscolari Spinali segmentali,
focali e distali, la Neuropatia Motoria Multifocale (Multifocal Motor
Neuropathy, MMN), l'Atrofia Muscolare Bulbospinale Progressiva o Malattia di
Kennedy (Progressive ulbospinal Muscular Atrophy, PBSMA), la
Sindrome di Brown-Vialetto-Van Laere (Paralisi pontobulbare con sordità
sensoriale), l'Atrofia Muscolare Spinale (Spinal Muscolar Atrophy, SMA)
correlata con mutazioni del gene smn e la SMA familiare progressiva a esordio
tardivo, la Paraplegia Spastica Ereditaria o Malattia di Strùmpel-Lorrain
(Hereditary Spastic Paraplegia, HSP), la Sindrome di Fazio-Londe(Paralisi
infantile bulbare progressiva), e altre forme patologiche e eziopatogenesi
tossica, attinica o infettiva - come la Sindrome PostPolio, che si verifica in
soggetti con pregressa poliomielite - : clinicamente, peraltro, i termini SLA e
MND vengono considerati sinonimi, a indicare una patologia rapidamente
progressiva caratterizzata da paralisi, ipertrofia, atrofia muscolare,
fascicolazioni e crampi.
approfondimenti : http://www.drbensi.it/persapernedipiu.htm
Il deficit neurologico
Una condizione che implica uno stato di sofferenza delle vie nervose motorie o
sensitive.
La via nervosa motoria percorre il Sistema Nervoso Centrale rappresentato
dall'asse cerebrospinale e il Sistema Nervoso Periferico rappresentato dai nervi
e loro terminazioni.
Il Sistema Nervoso comprende il Sistema Nervoso Autonomo che innerva gli organi
vitali, cuore polmoni e ghiandole e controlla i muscoli involontari.
MOTILITA'
L'origine del movimento
Esiste una rappresentazione corticale della motilità nell'area 4 o area motoria
primaria della circonvoluzione precentrale a livello dello strato delle cellule
piramidali. Inoltre un altro fascio di fibre deriva dai motoneuroni dell'area 6
(posta anteriormente all'area motoria primaria) e dalle aree della corteccia
retrorolandica (3,1,2).
Tutte queste fibre che originano dai neuroni motori corticali confluiscono a
formare la via piramidale o via motoria primaria. Il neurone motorio è detto
primo motoneurone.
LA VIA MOTORIA PRIMARIA
Cenni
E' una via neuronale semplice, incrociata, con due sole stazioni (sinapsi), una
a livello delle corna anteriori del midollo spinale e l'altra periferica, a
livello della giunzione neuromuscolare.
Come si propaga l'impulso nervoso dal centro
alla periferia
Mentre la trasmissione di un impulso lungo una fibra nervosa è un fenomeno
elettrico, il passaggio dell'impulso da un motoneurone superiore
all'alfa-motoneurone, oppure da una alfa-motoneurone al muscolo (nella giunzione
neuromuscolare), è mediato chimicamente.
A livello delle sinapsi, quando tutto funziona a perfezione, si ha la
trasformazione di un fenomeno elettrico in un fenomeno chimico e nuovamente in
fenomeno elettrico con le stesse caratteristiche di segnale del primo.
Il controllo del movimento
Vi fanno parte oltre al sistema piramidale altre strutture neuronali, che
regolano la motilità, garantendone nell'insieme la plasticità, cioè tutto quell'insieme
di proprietà che consente al movimento di essere raffinato, graduato, selettivo,
finalistico etc.
I meccanismi sovraspinali di controllo del movimento
Vi sono dei sistemi di controllo sovraspinali che comprendono:
1) Il Sistema reticolare (via reticolo spinale) che nasce dalla
sostanza reticolare e che opera con un meccanismo di regolazione a feed-back
negativo, bloccando cioè una ulteriore contrazione muscolare quando questa è
elevata.
2) Il Sistema di controllo
extrapiramidale ha i suoi nuclei di origine nei gangli della base.
E' un sistema a doppio controllo, pertanto sia con funzione eccitatoria
(mediatore: dopamina) che inibitoria, con percorso finale di uscita in parte
sulla via reticolo-spinale e in parte sulla via cortico-spinale. Le patologie
che interessano questo sistema sono responsabile di Sindromi Parkinsoniane.
3) L'ultimo sistema di controllo
sovraspinale della motricità è quello del cervelletto. E' un sistema assai
complesso e molto importante nel garantire raffinatezza e precisione del
movimento, oltre che fondamentale nel regolare la statica, l'equilibrio e i
rapporti spaziali del corpo.
I meccanismi spinali di controllo del
movimento
A livello spinale un primo controllo è attuato dalla cellula di Renshaw, che è
un interneurone inibitorio di connessione tra la sinapsi del motoneurone
piramidale e il motoneurone alfa. Il mediatore chimico è il GABA. Si dice
inibitorio perché serve a diminuire la frequenza di scarica delle cellule
corticali, allorché sia necessario regolarla in funzione dello sforzo da
compiere: devono essere attivati solo gli alfa motoneuroni utili a far compiere
al muscolo una contrazione efficace ma misurata.
Un altro sistema di controllo è il riflesso miotatico, che serve per la
regolazione del tono posturale.
Il tono posturale cambia a seconda della posizione del nostro corpo e si annulla
se si è sdraiati.
E' un riflesso dipendente dai fusi neuromuscolari contenuti nel muscolo, le cui
fibre si attivano automaticamente. Rapportandosi sempre con gli alfa motoneuroni
ne regolano la scarica, che modifica la contrazione di quel muscolo al fine di
mantenere costante il tono posturale.
PATOLOGIA DELLA VIA MOTORIA PRIMARIA
Cenni
Aspetti clinici delle diverse forme di lesioni a livello della via piramidale.
Le lesioni a livello della via piramidale Provocano vari gradi di riduzione
della forza muscolare o paresi, fino alla paralisi completa (o plegia).
Emiparesi ed emiplegia
Ad una lesione cerebrale, al di sopra dell'incrocio delle fibre piramidali,
segue dunque emiparesi o emiplegia della parte controlaterale del corpo.
Solo a seguito di lesioni che interessano il fascio genicolato, seguono paresi o
plegie omolaterali (dallo stesso lato) limitate alla faccia.
Tipica delle lesioni piramidali è la spasticità
Come si manifesta la spasticità?
Con un aumento del tono (ipertonia) dei muscoli antigravitari, flessori degli
arti superiori ed estensori degli arti inferiori. Si accompagna ad una esagerata
risposta al colpo del martelletto dell'esaminatore, nell'evocare un riflesso
propriocettivo, ad esempio colpendo il tendine rotuleo al ginocchio (iperriflessia).
Non esiste un solo tipo di risposta patologica a questa manovra, ma è possibile
una variabilità di risposte graduate che si pongono alla valutazione dell'occhio
esperto dello specialista esaminatore (riflessi policinetici, clono esauribile o
non esauribile etc.).
Si instaura allorché viene meno il controllo sovraspinale della motricità
I riflessi patologici: il BABINSKY
Si tratta di un riflesso patologico esterocettivo,
normalmente assente nel soggetto sano.
Nei pazienti con sindrome piramidale, compare strisciando con un oggetto a punta
stondata la pianta del piede in direzione calcagno-dita e si manifesta, al
contrario di quanto avviene nel normale, con l'estensione dell'alluce.
Le dita possono seguire o meno l'alluce in una apertura a ventaglio.
A volte una reazione in estensione dell'alluce si ottiene anche con una
pressione più o meno forte, limitata alla pianta del piede. Non sempre le
risposte sono chiare, anche quando viene aumentata progressivamente la forza
strisciando sul piede. L'incerto significato patologico non deve, tuttavia, far
trascurare la possibilità di trovarci di fronte ad una malattia del sistema
nervoso centrale.
Il Babinsky è costantemente presente nelle
sindromi piramidali midollari?
No. Il segno di Babinsky può mancare anche quando l'accentuazione dei riflessi è
molto marcata, tuttavia allorché è presente è sempre indicativo di una lesione
piramidale.
LESIONI DEL SECONDO MOTONEURONE
L'interruzione della via motoria a livello delle corna anteriori del
midollo, provoca paresi o paralisi omolaterale.
Dal secondo motoneurone dipende il contributo trofico per il muscolo.
Quando questo viene a mancare, il muscolo si indebolisce, perde tono, diminuisce
di volume, si atrofizza.
Questo processo si instaura nel tempo, per cui inizialmente il deficit del
trofismo è sfumato e così pure una diminuzione dei riflessi propriocettivi.
I segni di denervazione
Vi sono fenomeni positivi messi in atto dal muscolo nel tentativo di sopperire
ad un danno nervoso, come le fibrillazioni (valutabili solo con esame
elettromiografico) e le fascicolazioni, visibili, come piccoli scatti o
vibrazioni della pelle: sono segni peculiari di sofferenza dell'alfa
motoneurone. Questi segni, detti di denervazione, cessano allorché si stabilizza
una atrofia neurogena totale.
Poiché ogni fibra muscolare è innervata da più motoneuroni alfa appartenenti a
2, 3 radici spinali, per avere un muscolo totalmente paretico, la lesione deve
necessariamente essere pluriradicolare.
Le radici spinali confluiscono a formare i plessi
nervosi come il plesso brachiale e il plesso lombo-sacrale e quindi il nervo
periferico.
Funzione dei plessi nervosi
I plessi nervosi hanno una funzione specifica, simile a quella esplicata dalla
diramazione fascicolare del nervo anatomico, vale a dire quella di proteggere da
un eventuale denervazione dovuta a lesione di una singola porzione del plesso;
attraverso la complicata struttura del plesso le fibre nervose passano da un
determinato segmento di midollo spinale a numerosi e differenti muscoli.
L'innervazione di un singolo muscolo proviene normalmente da più di un segmento
di midollo spinale.
Dermatomeri e miomeri
L'area di pelle innervata da un singolo elemento di midollo spinale viene
denominata dermatoro; allo stesso modo i muscoli riforniti da un singolo
segmento di midollo spinale sono denominati miomeri.
La conoscenza di queste rappresentazioni segmentarie, dei muscoli e delle aree
di pelle innervate da determinati nervi, sarà determinante per porre una
diagnosi di compromissione di un nervo periferico o di una radice.
Il medico esperto riconoscerà immediatamente che la debolezza dei tricipiti e
degli estensori del polso e delle dita non può essere imputata alla lesione di
C7-C8, bensì ad una paralisi del nervo radiale, dal momento che è interessato
anche il brachioradiale.
LESIONI DELLA GIUNZIONE NEUROMUSCOLARE
Vi è un blocco a livello della placca motrice
dell'impulso nervoso che pertanto non può raggiungere il muscolo. I sintomi sono
la faticabilità e la mancanza di forza che si accentuano col movimento.
Non c'è né diminuizione del tono, né diminuizione di volume del muscolo.
Sono dovute a patologia della giunzione sia la miastenia gravis che le sindromi
miasteniche paraneoplastiche. Nella miastenia il mediatore chimico, acetilcolina,
trova i recettori deputati al contatto già occupati da autoanticorpi anti
acetilcolina. Ne consegue un blocco della trasmissione.
LESIONI MUSCOLARI
Miositi e Distrofie muscolari sono caratterizzate
da una perdita di forza muscolare, ipotrofia e ipotonia muscolare, risultato di
una distruzione delle fibre muscolari e nervose ad esse interposte ed essenziali
per il funzionamento dei fusi muscolari.
ALCUNI CENTRI DIAGNOSTICI
Università Cattolica Policlinico Gemelli Istituto di Neurologia
06.30154435
06.35501909
ptonali@rm.unicatt.it
Largo Gemelli, 8, 00168, ROMA
http://www.rm.unicatt.it/
Azienda Policlinico Umberto 1°
V.le del Policlinico, 155
V.le dell'Università, 39
V.le Regina Elena, 324
Via Lancisi
00161, ROMA
per informazioni telefoniche rivolgersi al numero
06.49970900 dalle ore 8.00 alle ore 18.00
policlinico@tuopoliclinico.roma.it
http://www.policlinicoumberto1.it/
Istituto Nazionale Neurologico "C. Besta" prof. Zeviani
02.2394388
02.2664236
zeviani@tin.it
Via Celoria 11, 20136, MILANO
http://www.mitopedia.org/home.htm
http://www.istituto-besta.it/#
Università degli Studi di Pisa Dipartimento di Neuroscienze Centro Miopatie
050.993046
050.554808
gsicilia@med.unipi.it
Via Roma 67, 56126, Pisa
http://www.med.unipi.it/neuroscienze/
Universita' di Pavia - Policlinico S.
Matteo - Dip. Patologia Umana ed Ereditaria
Via Forlanini 14 - 27100 - PAVIA
Tel. 0382.503429
Fax 0382.525866
http://www.saninlinea.net/site/924-0-0.htm
Universita' degli Studi di Verona Policlinico Borgo Roma - Dip. Scienze
Neurologiche e della Visione -
Tel.045.8071111
Piazzale L.A. Scuro,
10 - 37134 Verona
http://www.ospedaliverona.it/portale/default_fla.asp
Universita' degli Studi di Siena
Policlinico Le Scotte - Ist. Scienze Neurologiche - Lab. Genetica Molecolare per
le Malattie Neuromuscolari
Viale Bracci - 53100 - SIENA
Tel. 0577/585760-3
Fax 0577/40327
http://www.ao-siena.toscana.it/
Università degli Studi di Milano Istituto di
Clinica Neurologica
Te.0255033817
Tel.0255190392
gpcomi@mailserver.unimi.it
Giacomo.Comi@unimi.it
Nereo.Bresolin@unimi.it
Via F. Sforza, 35, 20122, MILANO
http://www.centrodinoferrari.com/
Associazioni :
UILDM Unione Italiana Lotta Distrofia Muscolare
0498021001
049757033
direzionenazionale@uildm.it, Via Vergerio, 19/2, 35126, PADOVA
http://www.uildm.org
|

