|
Per ottenere un gene devo partire da un tessuto, un
organo o un ammasso di cellule, da questi posso avere tre cose:
. DNA,
. RNA totali (da cui posso ricavare l'RNA messagero),
. proteine totali (proteina purificata x -tramite uso di anticorpi
per il suo riconoscimento-).
Conoscendo le zone fiancheggianti al gene di interesse posso costruirmi
un oligonucleotide e isolare il mio gene. Posso anche derivarlo
partendo dal gene omologo a quello cercato, ma presente (e identificato)
su un'altra specie: la PCR sarà svolta in condizioni non troppo
strette di annealing.
Volendo posso però costruirmi delle banche, o delle genoteche: ossia
avere tutta una serie di cloni ricombinanti che sono rappresentativi
dellintero genoma di un certo organismo, in questo modo ho tutto
il genoma di un organismo clonato in opportuni vettori.
Ci sono 2 tipi di banche, a seconda che il genoma di partenza abbia
o no gli introni.
Senza introni estraggo il DNA genomico e costruisco banche genomiche,
se ho introni (mammifero) estraggo gli RNA totali, isolo i messaggeri
e ricavo con la trascrittasi inversa il cDNA, con questo costruisco
una banca a cDNA.
Una volta che ho trasformato Coli, e ho il materiale su cui lavorare,
un po di colonie trasformate contenenti il gene clonato le uso
per caratterizzare il gene (screening) e in parte amplifico la cultura
e la conservo a 80°C (storage).
Passando attraverso il cDNA non ho più una rappresentitività numerica
dei geni della cellula, ma ho un arricchimento: certi geni sono
espressi più di altri, e questi saranno amplificati di più, a seconda
del tessuto e delle condizioni originarie del tessuto prima che
arrivasse in laboratorio.
Per screenare le banche ci sono diversi metodi, il punto di partenza
è che devo avere una sonda specifica per quello che vado a marcare.
In entrambi i casi io arrivo allisolamento del gene. Nel caso in
cui avessi solo la proteina purificata esistono metodi di sequenziamento
delle proteine da cui posso costruirmi un gene sintetico, tenendo
conto di tutti i fattori (sonde di Guessmer: oligo sonda di circa
20nucleotidi - circa 7 amminoacidi- che tiene conto del codice degenerato,
scegliendo una regione proteica che contenga il numero minimo di
codoni degenerati
), oppure posso farmi degli anticorpi, che mi servono per screenare
le banche ad espressione.
Ottenuto il gene, lo sequenzio e ci posso fare tutti gli studi
che voglio: studiare il 3, studiare il 5, studiare comè regolato
durante il ciclo, studiare quando è espresso, ecc., con vari passaggi
diversi a seconda del tipo di biotecnologia (industriale, farmaceutica
).
In tutti i casi quello che interessa a tutti è, dopo aver isolato
il gene, produrre in grosse quantità la proteina (così la purifico
bene e la studio) e a seconda del prodotto potrò avere trials clinici
o processi migliorativi industriali.
Per allestire le librerie genomiche dal cDNA parto dalle cellule
e arrivo alla scelta del vettore (in funzione della dimensione dei
frammenti genici),
estraggo il DNA e lo digerisco (così comè non può essere clonato
108bp!), stando attento che la banca che vado a costruire
sia rappresentativa di tutto il genoma, frammentando il meno possibile
tra i geni (digestioni parziali seriali).
Una volta che lho digerito posso o clonarlo tutto o selezionare
solo frammenti di alcune dimensioni (size fractionation). Quindi
clono il tutto.
E ovvio che nella banca a cDNA non devo frammentare i messaggeri!
In questi casi i cDNA non hanno estremità con siti di restrizione,
quindi devo aggiungo dei linker (un po come la PCR), ma prima di
fare questo devo metilare il cDNA, altrimenti mi si spezzetta tutto,
infatti io non conosco la sequenza e non conosco la mappa di restrizione.
Una volta clonati o trasformo Coli coi plasmidi o faccio il packaging
con i fagi.
Il passaggio successivo è la caratterizzazione della banca: devo
sapere veramente cosa cè nei miei vettori percui una volta trasformato
estraggo il DNA ricombinante da Coli e lo caratterizzo (ottengo
la mappa di restrizione per avere unindicazione delle dimensioni).
Ora devo titolare la banca: devo sapere, usando un certo quantitativo
di dna e di cellule di coli quanti cloni ricombinanti ottengo (devo
sapere la resa). Perché? perché devo sapere quando screeno un gene
quanti cloni ricombinanti devo analizzare per essere sicuro di trovare
il gene che più mi interessa (più il genoma è complesso più ho cloni
da analizzare) e per avere la probabilità dalla mia parte ne analizzarò
un po di più. Questo vale anche per i frammenti: più sono piccoli
e più analisi devo fare.
Ora da una parte faccio lo screening, dallaltro lo storage (con
tanto di amplificazione di quello che mi rimane: se ho fagi, ne
uso un po per trasformare Coli, se ho i plasmidi, faccio crescere
coli, piastro , raccolgo e metto via), In questii passaggi di amplificazione
devo pure tener conto del fatto che i geni esogeni potrebbero essere
tossici per lospite. Magari al primo passaggio lospite conserva
lesoDNA, ma successivamente tende a perderlo. Devo stare molto
attento: potrei arricchire certi geni o perderli.
Cè una formula, derivata dalla legge di Poisson, che mi permette
di calcolare il n° di cloni che io devo analizzare per avere una
certa probabilità di trovare il gene di mio interesse. Il numero
di cloni da analizzare cresce con:
1. la probabilità che io voglio di trovare il gene di mio interesse;
2. la complessità del genoma dellorganismo di cui ha fatto la banca;
3. la piccolezza dei frammenti di DNA clonati nellorganismo ospite
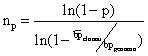 Nel
caso del genoma umano (3x109 bp), ammettendo di avere
dimensioni medie dei frammenti clonati 2x104, e ammettendo
di volrere la probabilità del 90% di trovare il frammento x, il
risultato è che io devo screenare almeno 690.000 cloni, isolati
ed indipendenti. Immaginare un po quante piastre bisogna preparare! Nel
caso del genoma umano (3x109 bp), ammettendo di avere
dimensioni medie dei frammenti clonati 2x104, e ammettendo
di volrere la probabilità del 90% di trovare il frammento x, il
risultato è che io devo screenare almeno 690.000 cloni, isolati
ed indipendenti. Immaginare un po quante piastre bisogna preparare!
Cè pure una formula un po più veloce ed empirica: il numero di
cloni da saggiare è uguale a 5 per il rapporto fra le dimensioni
del genoma e le dimensioni dei frammenti.
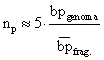 Devo costruire
la mia banca cercando di rappresentare tutto il genoma. La miscela
deve essere casuale(metodo non riproducibile), quindi non devo tagliare
favorendo frammenti grandi o piccoli, ed inoltre devo donar loro
estremità coesive. Uso allora enzimi di restrizione di tipoII.
Questi enzimi riconoscono 4, 6, 8, n bp, se ne uso uno da n, mi
darà 4n paia di basi per frammento. Teniamo conto che
non devono essere troppo piccolo e che i vettori hanno un limite. Devo costruire
la mia banca cercando di rappresentare tutto il genoma. La miscela
deve essere casuale(metodo non riproducibile), quindi non devo tagliare
favorendo frammenti grandi o piccoli, ed inoltre devo donar loro
estremità coesive. Uso allora enzimi di restrizione di tipoII.
Questi enzimi riconoscono 4, 6, 8, n bp, se ne uso uno da n, mi
darà 4n paia di basi per frammento. Teniamo conto che
non devono essere troppo piccolo e che i vettori hanno un limite.
In genere si preferiscono quelli che riconoscono 4 bp con digestioni
parziali (la parziale è necessaria), inoltre mi dà una distruibuzione
più casuale (ho più possibilità di configurazione). Se facessi digestioni
totali taglierei pezzetti di un gene in cloni diversi, e come faccio
a ricostruire il tutto? Invece con le parziali posso fare il chromosome
walking: tutti i siti non verranno tagliati con la stessa velocità
(uso [limitanti] o scarsità di tempo), il tutto è molto riproducibile,
perché il taglio dipende dallintorno del sito di restrizione (-->
overlapping), che è sempre lo stesso.
Chromosome Walking
Impiega un fago ricombinante dal quale isolare un altro fago che
contenga informazioni relative al genoma sovrapponibili (overlap).
Basta preparare due sonde derivate una dall’estremo 5’
e l’altra dall’estremo 3’ di questo primo coli
e ripetere con queste sonde lo screening della genoteca.
Si otterranno altri cloni.
Se ne sceglieranno due tra quelli diversi dal primo, uno più
spostato al 3’; l’altro più spostato al 5’.
Dal clone “3’” si prepara una sonda presente al
suo estremo 3’, e dal clone 5’ una sonda presente al
suo 5’. e pronti per un altro screening e così via,
camminando sul cromosoma.
Limiti. I piccoli segmenti di DNA utilizzati per vagliare di nuovo
la genoteca devono essere rappresentati una sola volta nel genoma:
ci sono problemi col DNA ripetuto, il rischio è di saltare
da un cromosoma all’altro.
|




