 Equilibrio CO2 - H2O
Equilibrio CO2 - H2O
Y.Quinif, "La corrosione per miscela di acque",
Speleologia, n. 5 (1981) p. 40-43.
A.A.Cigna, "Sulla classificazione dei fenomeni carsici",
Le Grotte d'Italie, 11 (1983) p. 497-505
M. Chiesi, "Il sistema calcite-gesso", Ipoantropo (1984), p. 29-30
P.Forti, "Processi ipercarsici e speleogenesi",
Speleologia 24 (1991) p. 42-46
P.Forti, "Processi ipercarsici e speleogenesi",
Speleologia 26 (1992) p. 11-15
A. Cigna, "Some remarks on phase equilibria of evaporites and other
karstifiable rocks", Le Grotte d'Italia, 12 (1986) 201-208
A.I. Pechorkin, "On gypsum and anhydrite distribution in zines near
to the surface of sulphate missifs", Le Grotte d'Italia, 12 (1986) 397-406
G. Badino, M. Meneghel, "Le grotte nei ghiacci dell'Antartide",
Speleologia, 43 (2000) 52-58
La classificazione dei fenomeni carsici basata sul numero delle componenti conivolte nel processo carsico distingue fenomeni di carsismo (a tre componenti), ipocrasismo (con meno di tre componenti) e ipercarsismo (piu` di tre). Infatti il caso piu` comune delle grotte nel calcare coinvolge tre componenti: calcare, anidride carbonica ed acqua.
Esempi di processi ipocarsici (con meno di tre componenti) sono:
Quando invece sono coinvolti piu` di tre componenti si parla di ipercarsismo. Questi fenomeni sono comunemente ascrivibili ad ambienti idrotermali ma possono avere rilevanza anche in situazioni "normali".
In genere i processi ipercarsici coinvolgono altri sali oltre alla CaCO3, che intervengono nella chimica del processo carsico. Questi sali possono avere uno ione in comune con la sostanza carsificata, oppure intervengono solo indirettamente influenzando la forza ionica della soluzione.
In generale l'aggiunta di un sale con uno ione in comune con il soluto in soluzione diminuisce la solubilita di questo. Tuttavia cio` non e` sempre vero: l'aggiunta di carbonato di magnesio aumenta la solubilita` della calcite (effetto Picknett). Questa e` la ragione dei grandi vuoti al contatto fra calcare e dolomia.
La solubilita` della CaCO3 cresce al crescere della salinita`
(forza ionica). Ad elevata salinita` (-7, come nel mare) la solubilita`
di CaCO3 supera 210 ppm, indipendentemente dalla concentrazione
della CO2 nell'atmosfera.
A basse salinita` (0, come nell'acqua piovana) la solubilita` dipende
invece dalla CO2 ed e` dell'ordine di 100 ppm.
Valori tipici dei sali disciolti:
La diminuzione della solubilita della sostanza gia` presente, ne provoca la precipitazione. Pertanto si possono avere sia fenomeni di accentuata corrosione che fenomeni di deposizione, di concrezionamento e mineralizzazione.
L'ipercarsismo termale e` dovuto alla miscelazione di acque calde in risalita con acque fredde di origine meteorica.
Il carsismo dipende essenzialmente dalla corrosione della roccia (sovente calcare, percio` principalmente carbonato di calcio, CaCO3) da parte delle acque. L'acqua pura scioglie poco il carbonato di calcio (16 mg/l). Pero` l'acqua in contatto con l'atmosfera contiene disciolta una piccola quantita` di CO2 che e` sufficiente ad aumentare la solubilita` del carbonato ci calcio (70 mg/l a 0oC, e 50 mg/l a 15oC). Nel suolo, sotto una copertura vegetale o sotto la neve, l'acqua si arricchisce ancor piu` di anidride carbonica, e puo` disciogliere anche alcune centinaia di mg/l di carbonato di calcio. La tabella sotto riporta lacuni valori tipici delle concentrazioni di CaCO3 disciolta in acqua.
| CaCO3 disciolta in acqua | |
| Alta montagna | 100 mg/l |
| Regioni temperate | 240 mg/l |
| Regioni mediterranee | 200 mg/l |
| Regioni intertropicali | 170 mg/l |
Nelle regioni fredde il gelo inibisce la corrosione carsica superficiale per parecchi mesi l'anno. Pero` sotto la neve si formano sacche d'aria ricche di anidride carbonica che rende l'acqua di fusione piu` aggressiva. In clima temperato la corrosione e` frenata dall'aumento della temperatura, ma e` favorita dalla presenza di vegetazione. Nelle regioni calde e secche invece la corrosione superficiale e` ostacolata dalla alta temperatura e dalla scarsita` di copertura vegetale. In queste regioni pero` la corrosione profonda e` possibile poiche` le acque di condensazione sono molto piu` fredde sotto terra che in superficie. Infine nelle regioni calde tropicali la corrosione e` intensa tanto in superficie quanto in profondita`, a causa principalmente della vegetazione.
Equilibrio CO2 - H2O
La quantita` di anidride carbonica disciolta in acqua e` proporzionale alla pressione parziale di essa nell'atmosfera (legge di Henry)
dove il coefficiente KH e` costante di Henry per la CO2. La frazione di anidride carbonica disciolta puo` essere espressa in litri per litro d'acqua o in milligrammi (il rapporto fra questi due vale 1.964 gr/l =44/22.4), secondo la tabella sotto riportata che contiene i coefficienti di solubilita` alla pressione di una atmosfera. Quando la pressione parziale della CO2 e` inferiore, la quantita` disciolta in acqua e` ottenuta moltiplicando questi coefficienti per la sua pressione parziale (per esempio in aria questa vale circa 0.0003 atm).
La percentuale di anidride carbonica nelle cavita` e` superiore. Il tasso di CO2 nel sottosuolo e nel reticolo di fratture vale tra 2 e 6 %. Nella zona delle gallerie aerate scende a 0.1 - 2 %. Nella zona profonda di dissoluzione ha valori generalmente compresi fra 0.2 e 4 %.
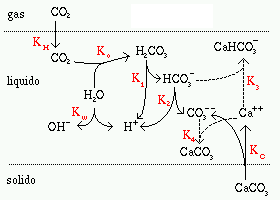
La soluzione di anidride carbonica in acqua provoca la formazione di ioni
carbonato e bicarbonato in soluzione.
CO32- con H+ secondo le reazioni
schematizzate in figura, le cui costanti di equilibrio sono
riportate nella tebella sotto.
 Si introduce la concentrazione effettiva della CO2 disciolta,
come somma di quella libera in soluzione e quella legata,
Si introduce la concentrazione effettiva della CO2 disciolta,
come somma di quella libera in soluzione e quella legata,
Il valore di 1/Ko e` piccolo, circa 0.003, pertanto solo lo 0.3% della CO2 in soluzione e` dovuta all'acido carbonico.
Le costanti di equilibrio delle reazioni sono espresse in termini delle attivita`, (x), dei reagenti. Queste sono legate alle concentrazioni, [x], tramite i coeffivienti di attivita` (x) = cx [x], i quali dipendono dalla forza ionica della soluzione,
secondo log(cx) = - A I1/2 Zx2 / ( 1 + B I1/2 ax ), dove ax e` il raggio ionico effettivo del reagente ( che vale 9 per [H+], 4.5 per [CO32-], 4 per [HCO3-], e 3 per [OH-] ). I coefficienti A e B valgono A=0.4883 + 8.074 10-4 T[oC] e B=0.3241 + 1.6 10-4 T[oC].
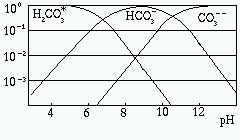
L'equilibrio di queste reazioni e` governato dal pH della soluzione oltre che dalla pressione della CO2 nell'atmosfera. La figura a lato riporta le frazioni molari di H2CO3*, HCO3- e CO32- rispetto alla concentrazione totale di atomi di carbonio. Da notare che per pH inferiori a 8.3 (come e` generalmente il caso per le acque carsiche), la frazione di [ CO32- ] e` trascurabile.
La cinetica di questo equilibrio e` dominata dalla reazione H2O + CO2 --> H2CO3, che e` abbastanza lenta. Tipicamente ci vogliono 50 secondi per arrivare al 50% dell'equilibrio e 100 sec. per il 99%.
Equilibrio CO2 - H2O - CaCO3
In soluzione acquosa il carbonato di calcio e` ionizzato CaCO3 -> Ca2+ + CO32- e la CO32- si combina con H+ per formare HCO3-. Si tratta di tre reazioni scrivibili globalmente
Il prodotto (solubilita`) delle concentrazioni dei due ioni all'equilibrio e` Kc = ( Ca2+ )eq ( CO32- )eq dipende solo dalla temperatura. I valori di Kc sono molto piccoli e decrescono leggermente con la temperatura. La saturazione rispetto al calcio e` il rapporto rispetto ad Kc delle effettive concentrazioni
Gli ioni CA2+ in soluzione reagiscono con HCO3- e con CO32-. Queste due reazioni hanno costanti d'equilibrio molto piccole e possono essere trascurate nelle condizioni dei carsi naturali.
La dissoluzione di Ca aumenta [ CO32- ] e diminuisce [ H+ ] e [ H2CO3* ], cioe` essa consuma CO2. L'evoluzione di queste concentrazioni dipende dal rapporto fra i volumi di atmosfera e liquido, X = Vg / Vaq . Quando l'acqua scorre nelle fessure senza atmosfera o nella zona allaganta (sistema chiuso) X=0. Quando scorre nelle gallerie il sistema e` aperto e X e` molto grande.
L'equilibrio chimico e` governato dalle leggi di massa (tabella sotto), dalla condizione di neutralita` elettrica,
(che essenzialmente si riduce a 2 [Ca2+] = [HCO3-], e quindi la forza ionica e` circa I = 3 [Ca2+] ), dalla condizione di conservazione degli atomi di carbonio,
Da notare che [CO32-]i e` trascurabile, e [H+]i = [HCO3-]i per cui per la legge di massa, [HCO3-]i = sqrt( [H2CO3*]i K1 / cH,i cHCO3,i ). Queste equazioni messe assieme permettono di ottenere una relazione fra [Ca2+] e [H+], dopodiche` le concentrazioni delle altre specie ioniche possono essere ricavate. Queste relazioni descrivono l'andamento chimico della soluzione al variare della concentrazione di ioni calcio disciolti.
La cinetica delle reazioni chimiche e` dominata dalla reazione piu` lenta, percio` e` il passaggio in soluzione del carbonato di calcio che determina la velocita` d'insieme di questo sistema di equazioni chimiche. Il tasso di dissoluzione "teorico" alla superficie e` dato dalla somma delle attivita` moltiplicate per le costanti cinetiche delle reazioni dirette meno quella inversa (equazione PWP, di Plummer, Wigley Parkhurst)
Questa equazioni pero` da' valori troppo grandi, di un ordine di grandezza, perche` non tiene conto della diffusione degli ioni nell'acqua e del ruolo della CO2.
 Il tasso di dissoluzione alla superficie e` proporzionale ad una funzione
della differenza fra la concentrazione degli ioni alla superficie,
cs, e quella di equilibrio:
Fs = as f( ceq - cs ).
Questo deve essere pari alla diffusione nel liquido,
Fd = (D/h) ( cs - cl ),
dove D e` il coefficiente di diffusione (tipicamente 10-5
cm2/sec), h e` lo spessore
dello strato limite, e cl e la concentrazione
nel liquido.
La cinetica e` dominata dalla diffusione quando as e`
grande, e dalla dissoluzione alla superficie quando e` piccolo.
Il tasso di dissoluzione alla superficie e` proporzionale ad una funzione
della differenza fra la concentrazione degli ioni alla superficie,
cs, e quella di equilibrio:
Fs = as f( ceq - cs ).
Questo deve essere pari alla diffusione nel liquido,
Fd = (D/h) ( cs - cl ),
dove D e` il coefficiente di diffusione (tipicamente 10-5
cm2/sec), h e` lo spessore
dello strato limite, e cl e la concentrazione
nel liquido.
La cinetica e` dominata dalla diffusione quando as e`
grande, e dalla dissoluzione alla superficie quando e` piccolo.
Nel processo pero` interviene anche la conversione di CO2 in H+ e HCO3-, il cui tasso e` fCO2 A/V, dove A e` l'area della superficie e V e` il volume del liquido. Questo deve egualiare Fs. Quando il volume V e` molto piccolo questa e` la reazione determinante la cinetica.
In condizioni chiuse con flusso laminare (fessure e microfratture) il tipo di cinetica dipende dall'ampiezza H della fessura. Per H< 2 10-5 m la cinetica e` dominata dalla conversione di CO2 e la dissoluzione cresce linearmente con H. Per valori di H superiori cresce piu` lentamente ed la cinetica e` governata ssia dalla conversione di CO2 che dalla diffusione. Per H> 1 mm il tasso di dissoluzione resta costante e quando H supera 5 cm predomina solo la diffusione. Se il flusso e` turbolento (strato limite inferiore a 10-4 cm) la diffusione e` determinata dai vortici: in pretica il coefficiente di diffusione e` 104 volte piu` grande. La dissoluzione e` controllata dalla conversione di CO2. Per H> 1 mm il tasso di dissoluzione diventa costante e la cinetica e` controllata dalla reazione alla superficie.
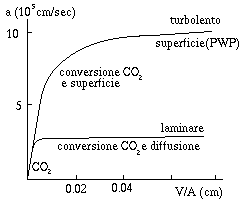
In generale il tasso di dissoluzione risulta proporzionale alla differenza fra la concentrazione e quella di equilibrio,
dove il coefficiente a dipende da temperatura, pressione parziale di CO2, rapporto V/A, spessore dello strato limite, e tipo di flusso. La figura a lato mostra il coefficiente a al variare di V/A per flusso laminare e turbolento (T = 10oC, PCO2 = 0.05 atm). La concentrazione di ioni calcio varia nel tempo dc/dt = (A/V) F(c). Poiche' F(c) e' lineare c cresce esponenzialmente verso il valore di equilibrio (la costante di tempo e' dell'ordine di 100 sec).
Queste formule sono in buon accordo coi dati sperimentali per concentrazioni abbbastanza lontane da ceq. Per valori prossimi alla concentrazione di equilibrio l'andamento di F(c) non e' piu' lineare (come secondo l'equazione PWP) ma ha un andamento del tipo
dove co rappresenta una soglia di transizione. I valori degli esponenti n e n' e dei coefficienti k e k' dipendono dalla roccia. In genere n'>n. La discrepanza dalla legge lineare (PWP, che comunque vale per la calcite sintetica ultrapura) e' dovuta alla presenza di impurita' che vengono adsorbite sulla superficie ed inibiscono la dissoluzione degli ioni calcio. Si puo' scrivere
dove T(c) e' l'isoterma di Fowker Frumkin (l'entalpia di adsorbimento cresce lineramente col grado di adsorbimento).
In conclusione per flussi turbolenti e grandi (V/A > 1 mm) la cinetica e' dominata dalle reazioni alla superficie e vale la legge PWP. Per flussi laminari in fessure inferiori al mm e per i veli sulle pareti la cinetica e' lineare e dominata della reazione H2O + CO2 = H2CO3. Per flussi laminari piu' grandi interviene anche la diffusione. Vicino all'equilibrio le reazioni superficiali diventano predominanti (a causa della inibizione).
| Reazione | K | (T in oK) | 0oC | 5oC | 10oC | 15oC | 20oC | 25oC | unit |
| H2O + CO2 -> H2CO3 | Ko = (CO2) / (H2CO3) = KH / K' | Ko = 330 circa
Ko = 1.7 10-4 / K1 |
5.54 | 5.54 | 5.64 | 5.60 | 5.53 | 5.56 | 101 |
| K' = [H2CO3] / PCO2 | 7.58 | 6.30 | 5.37 | 4.57 | 3.89 | 3.38 | 10-2 | ||
| H2CO3 -> H+ + HCO3- | (H+) (HCO3-) = K1 (H2CO3*) |
log(K1) = - 356.3094 - 0.06091964 T + 21834.37 / T - 126.8339 log(T) - 1684915 T-2 [M/l] | 2.24 | 2.45 | 2.69 | 2.95 | 3.23 | 3.54 [FIXME 4.3] |
10-7 M/l |
| HCO3- -> H+ + CO32- | (H+) (CO32-) = K2 (HCO3-) |
log(K2 = - 107.8871 - 0.03252849 T + 5151.79 / T + 38.92561 log(T) - 56371.9 T-2 [M/l] | .... | 2.88 | 3.23 | 3.71 | 4.17 | 4.68 [FIXME 4.4] |
10-11 M/l |
| CaCO3 -> Ca2+ + CO32- | Kc = (Ca2+) (CO32-) | log(Kc = - 171.9065 - 0.077993 T + 2839.319 / T + 71.595 log(T) [(M/l)2] | 3.63 | 3.71 | 3.47 | 3.23 | 3.02 | 2.81 | 10-9 (M/l)2 |
| CO2(g) -> CO2(aq) | KH = (CO2) / PCO2 | log(KH) = 108.3865 + 0.01985076 T - 6919.53 / T - 40.45154 log(T) + 669365 T-2 [M/l atm] | 1.71 | 1.42 | 1.19 | 1.02 | 0.88 | 0.76 | ml(CO2) / ml(H2O)
(riportati a 0oC) |
| M = KH 2.144 | 3.67 | 3.04 | 2.55 | 2.19 | 1.89 | 1.63 | gr / l(H2O) | ||
| H2O -> H+ + OH- | Kw = (H+) (OH-) | log(Kw = 22.801 - 0.010365 T - 4787.3 / T - 7.1321 log(T) [(M/l)2] | 1.15 | 1.82 | 2.88 | 4.57 | 7.07 | 10.96 | 10-15 (M/l)2 |
| K1 Kc / K2 H | .... | 3.58 | 2.75 | 2.09 | 1.64 | 1.29 | 10-8 | ||
| Ca2+ + HCO3- -> CaHCO3+ | K3 = (CaHCO3+) / (Ca2+) (HCO3-) | log(K3 = 1209.12 + 0.31294 T - 34765.05 / T - 478.782 log(T) [M/l] | |||||||
| Ca2+ + CO32- -> CaCO3 | K4 = (CaCO3) / (Ca2+) (CO32-) | log(K4 = -1228.732 - 0.299444 T + 35512.75 / T + 458.818 log(T) [M/l] |
[FIXME: Le espressioni riportate per Kw, Kc, e KH sembrano dare valori corretti. Quelle per K3 e K4 danno valori delle costanti troppo alti, mentre queste dovrebbero essere quasi nulle. Infine Quelli per K1 e K2 danno valori che non hanno senso.]
Nella tabella sopra riportata i logaritmi sono in base 10.
Il coefficiente di 1/T e` legato all'entalpia di reazione, HR, secondo l'equazione di van't Hoff (R e` la costante dei gas, pari a 0.082 l atm/M oK),
Il coefficiente 2.144 e` ( 44 / 22.4 ) * (273+25)/273.
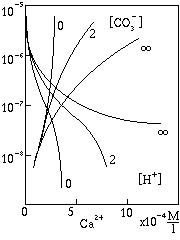 La variazione delle concentrazioni [H+] e
[CO32-] al variare della concentrazione di
Ca2+ in soluzione sono mostrate in figura, per differenti
valori di X, cioe` condizioni del sistema, da aperto (X
molto grande) a chiuso (X = 0).
La saturatione di calcio in soluzione varia in modo simile alla concentrazione
[CO32-].
[H2CO3*] decresce linearmente
con [Ca2+] con un coefficiente proporzionale a 1/X.
La variazione delle concentrazioni [H+] e
[CO32-] al variare della concentrazione di
Ca2+ in soluzione sono mostrate in figura, per differenti
valori di X, cioe` condizioni del sistema, da aperto (X
molto grande) a chiuso (X = 0).
La saturatione di calcio in soluzione varia in modo simile alla concentrazione
[CO32-].
[H2CO3*] decresce linearmente
con [Ca2+] con un coefficiente proporzionale a 1/X.
Nei sistemi carsici in cui l'acqua scorre in superficie prima di infiltrarsi nella roccia, la dissoluzione del calcio avviene in due modi: prima in condizioni aperte (lungo una linea quasi orizzontale nel diagramma [H2CO3*] - [Ca2+] ) poi in condizioni chiuse (lungo una linea molto inclinata). Il risultato e` equivalente ad una dissoluzione in condizioni chiuse con una maggiore pressione parziale iniziale di CO2. Questo spiega le alte concentrazioni di calcio (2 mM/l) in acque carsiche alimentate da un sistema diffuso.
Dalle equazioni del bilancio di massa e quella del bilancio della carica
(semplificata 2 [Ca2+] = [HCO3-] )
si ottiene la formula di Caro
per la concentrazione di equilibrio degli ioni H+
e calcio in soluzione in funzione della pressione
parziale della CO2:
Nei sistemi aperti PCO2 e` fissata e queste relazioni
permettono di ottenere la concentrazione d'equilibrio di Ca2+.
Nei sistemi chiusi e` fissata la PCO2 iniziale e quella finale
si esprime tramite essa e la concentrazione di calcio,
PCO2 = PCO2i - [Ca
L'influenza di ioni estranei sull'equilibrio della calcite ha differenti aspetti.
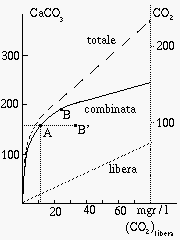 Nella CO2 in soluzione
(determinata dalla pressione di CO2 nell'atmosfera sovrastante),
si distinguono la porzione libera (in soluzione sotto forma di
acido carbonico)
e quella legata, combinata con i sali in soluzione perche` andata
a disciogliere il carbonato di calcio: la quantita` di carbonati
disciolti e` proporzionale alla quantita` di CO2 combinata,
in proporzione 2.273, pari al rapporto dei pesi molecolari,
100 e 44 rispettivamente.
All'equilibrio la soluzione non e` aggressiva.
Si ha una ben determinata quantita` di CO2 libera e
una di CO2
associata ai carbonati, e quindi un pH della soluzione ben preciso.
Dunque all'equilibrio c'e` una relazione che lega temperatura, pH,
e concentrazione di CaCO3- (v. tabella sotto e figura
a lato).
Nella CO2 in soluzione
(determinata dalla pressione di CO2 nell'atmosfera sovrastante),
si distinguono la porzione libera (in soluzione sotto forma di
acido carbonico)
e quella legata, combinata con i sali in soluzione perche` andata
a disciogliere il carbonato di calcio: la quantita` di carbonati
disciolti e` proporzionale alla quantita` di CO2 combinata,
in proporzione 2.273, pari al rapporto dei pesi molecolari,
100 e 44 rispettivamente.
All'equilibrio la soluzione non e` aggressiva.
Si ha una ben determinata quantita` di CO2 libera e
una di CO2
associata ai carbonati, e quindi un pH della soluzione ben preciso.
Dunque all'equilibrio c'e` una relazione che lega temperatura, pH,
e concentrazione di CaCO3- (v. tabella sotto e figura
a lato).
Se si discioglie una ulteriore quantita` di CO2 la soluzione diviene aggressiva: questo corrisponde ad un punto sotto la curva di equilibrio, punto B' in figura. La differenza B' - A rappresenta la CO2 aggressiva. Il pH della soluzione e` inferiore a quello di equilibrio. La soluzione si riporta allora all'equilibrio, aumentando sia la CO2 libera (e quindi il pH) che quella combinata, portandosi cioe` al punto B.
Per pH superiori al pH di equilibrio la soluzione e` satura di carbonati e si ha deposizione.
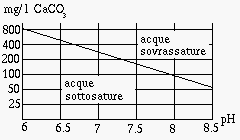 La misurazione del pH dell'acqua e della temperatura e` un indicatore della
aggressivita` dell'acqua.
Dalla misura della conducibilta` elettrica si risale alla concentrazione di
sali disciolti, che e` principalmente la CaCO3 disciolta
(tabella sotto riportata, oppure
abaco di Roques, in figura
e` riportata la curva per T=15OC).
Unitamente alla misura della pressione parziale della CO2
nell'atmosfera (cioe` alla sua percentuale) questo permette di valutare
se gli equilibri delle reazioni chimiche sono spostati a sinistra oppure
a destra e quindi se l'acqua e` aggressiva (corrode la roccia) oppure
se tende a depositare i sali in essa disciolti.
Se la [CO2] in soluzione (ottenuta dalla
P(CO2) con la legge di Henry) e` inferiore al valore riportato
si ha deposizione di carbonato di calcio. Se il valore e` superiore
si ha corrosione della roccia.
La misurazione del pH dell'acqua e della temperatura e` un indicatore della
aggressivita` dell'acqua.
Dalla misura della conducibilta` elettrica si risale alla concentrazione di
sali disciolti, che e` principalmente la CaCO3 disciolta
(tabella sotto riportata, oppure
abaco di Roques, in figura
e` riportata la curva per T=15OC).
Unitamente alla misura della pressione parziale della CO2
nell'atmosfera (cioe` alla sua percentuale) questo permette di valutare
se gli equilibri delle reazioni chimiche sono spostati a sinistra oppure
a destra e quindi se l'acqua e` aggressiva (corrode la roccia) oppure
se tende a depositare i sali in essa disciolti.
Se la [CO2] in soluzione (ottenuta dalla
P(CO2) con la legge di Henry) e` inferiore al valore riportato
si ha deposizione di carbonato di calcio. Se il valore e` superiore
si ha corrosione della roccia.
Nell'impiego dell'abaco di Roques bisogna tener presente i suoi limiti:
Il sistema di equazioni chimiche puo` essere impostato numericamente e risolto col calcolatore (Fritz, 1981, "Etude thermodynamique et modelisation des reactions hydrothermales e diagenetiques", Mem. 65 Sc. Geol. Univ. Strasbourg).
Il pH e` definito come l'antilogaritmo della concentrazione di ioni H+.
| CO2 libero (mgr/l) |
CaCO3 in soluzione (mgr/l) |
CO2 semicombinato (mgr/l) |
pH | CaCO3 in soluzione (mgr/l) |
CO2 semicombinato (mgr/l) |
pH |
| 0oC | 10oC | |||||
| 5 | 150 | 66.0 | 7.82 | 135 | 59.4 | 7.79 |
| 10 | 197 | 88.7 | 7.71 | 179 | 78.8 | 7.68 |
| 20 | 246 | 108 | 7.55 | 227 | 99.9 | 7.52 |
| 30 | 279 | 123 | 7.43 | 260 | 114 | 7.41 |
| 40 | 307 | 135 | 7.35 | 286 | 126 | 7.32 |
| 50 | 330 | 145 | 7.29 | 308 | 136 | 7.26 |
| 60 | 351 | 154 | 7.23 | 326 | 143 | 7.20 |
| 70 | 371 | 163 | 7.19 | 344 | 151 | 7.16 |
| 80 | 390 | 172 | 7.16 | 359 | 158 | 7.12 |
| 90 | 407 | 179 | 7.12 | 374 | 165 | 7.09 |
| 100 | 423 | 186 | 7.10 | 387 | 170 | 7.06 |
| 110 | 438 | 193 | 7.07 | 400 | 176 | 7.03 |
| 120 | 412 | 181 | 7.00 | |||
| 130 | 423 | 186 | 6.98 | |||
| 140 | 434 | 191 | 6.96 | |||
| 150 | 445 | 196 | 6.94 | |||
Ricapitolando: la solubilita` della CO2 diminuisce con la temperatura, mentre quella dalla CaCO3 aumenta.
L'aggressivita` dell'acqua cioe` la capacita` di sciogliere il calcare (calcite e dolomite) dipende dalla concentrazione di CO2. L'acqua di origine meteorica che passa nel terreno puo` arricchirsi di CO2 e diventare aggressiva. Questo succede se l'acqua attraversa terreni ricchi di acidi organici e in condizioni relativamente fredde, percio` gli ambienti piu` favorevoli allo sviluppo del carsismo sono quelli coperte di vegetazione e con clima temperato.
Pero` l'acqua di infiltrazione satura di CO2 diventa ben presto satura di carbonato, quindi il suo effetto corrosivo e` pressoche` limitato alla zona superficiale. La concentrazione della CaCO3 disciolta nell'acqua in un sistema carsico cresce andando dalle zone di assorbimento fino alle risorgenze. Essa varia a seconda del regime idrico che ha regolato il trasferimento dell'acqua e delle condizioni di scambio con la roccia: fessure e vaschette con flussi lenti hanno acque molto sature, condotti con flussi veloci portano acque con minor saturazione.
La corrosione dei massicci nelle zone profonde ha luogo poiche` le acque delle piene arrivano in profondita` in condizioni sottosature a causa del veloce trasferimento all'interno del sistema.
La miscelazione di acque sature puo` produrre acqua sottosatura, e quindi rinnovarne l'aggresivita` (iquesto e` chiamato effetto Bogli, anche se in effetti e` stato proposto da Laptev trent'anni prima). Questo effetto, certamente importante per acque marine, e` comunque debole per le acque dolci. La corrosione per miscela si realizza in assenza di atmosfera, altrimenti la CO2 si riequilibra con quella dell'aria. Quindi questo meccanismo si applica a zone allagate e/o sature (reticolo saturo). Le morfologie associate alla corrosione per miscelazione sono slarghi dopo una confluenza e cupole alimentate da canali di piccole dimensioni.
Quando l'acqua diviene sovrassatura si ha precipitazione di carbonato di calcio. All'interno della grotta questo da` luogo agli svariati fenomeni di concrezionamento. Quando la precipitazione avviene alle risorgenze si generano tufi e travertini.
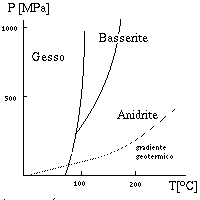 La solubilita` del solfato di calcio e` piu` alta di un ordine di grandezza
di quella del calcare (circa 2000 mgr/l per l'anidrite, e 2500 mgr/l per
il gesso; leggermente crescenti con la temperatura).
Il gesso e` la forma idratata ed e` piu` stabile a temperatura e pressione
normali.
L'anidrite predomina ad alte pressioni e/o temperature.
Percio` oltre una profondita` di circa 450 m si trova principalmente
anidrite.
La conversione di anidrite in gesso avviene durante l'emersione.
In essa il volume molare aumenta (di 1.626) ma il volume globale
diminuisce (teoricamente del 8.6%, in pratica di circa il 3%).
Il processo di idratazione dipende dalla struttura della roccia,
dal chimismo dell'acqua e da pressione e temperatura.
Si presume che avvenga sia per dissoluzione/precipitazione che per diffusione
di acqua nel reticolo cristallino.
La solubilita` del solfato di calcio e` piu` alta di un ordine di grandezza
di quella del calcare (circa 2000 mgr/l per l'anidrite, e 2500 mgr/l per
il gesso; leggermente crescenti con la temperatura).
Il gesso e` la forma idratata ed e` piu` stabile a temperatura e pressione
normali.
L'anidrite predomina ad alte pressioni e/o temperature.
Percio` oltre una profondita` di circa 450 m si trova principalmente
anidrite.
La conversione di anidrite in gesso avviene durante l'emersione.
In essa il volume molare aumenta (di 1.626) ma il volume globale
diminuisce (teoricamente del 8.6%, in pratica di circa il 3%).
Il processo di idratazione dipende dalla struttura della roccia,
dal chimismo dell'acqua e da pressione e temperatura.
Si presume che avvenga sia per dissoluzione/precipitazione che per diffusione
di acqua nel reticolo cristallino.
La reazione di dissoluzione del gesso e`
Il pK di questa reazione vale 4.667 - 5.197 10-9 T + 1.133 10-4 T2 (dove T e` in oC). L'indice di saturazione e` il rapporto fra le attivita` degli ioni calcio e solfato e la costante di equilibrio,
E` inferiore ad uno se la soluzione e` sottosatura, e maggiore di uno se e` sovrassatura.
La solubilta` varia con la pressione sulla roccia: 1/S dS/dP vale circa 400 Pa-1 per l'anidrite e 40 Pa-1 per il gesso. Il gesso presenta differente solubilita` a seconda delle dimensioni dei cristalli, con un massimo per 0.2-0.5 mm (per l'anidrite il massimo e` a circa 2.8 mm). La presenza di ioni estranei aumenta la solubilita`, per l'aumento della forza ionica della soluzione. La presenza di uno ione comune diminuisce la solubilita` di entrambi (come nel caso di CaCo3).
La cinetica del carsismo del gesso e` diminata dalla diffusione,
per il gesso e` del primo ordine (n e` circa uno), per l'anidrite del secondo ordine (n circa due). Il coefficiente k dipende dalle condizione dello strato limite (tipo di flusso, velocita` del flusso), proprieta` del minerale, rugosita` superficiale, presenmza di altri ioni, temperatura.
Sistema calcite gesso
Il gesso CaSO4.2H2O e il carbonato di calcio CaCO3 formano un sistema chimico con lo ione calcio Ca++ in comune. Questo sistema comprende anche la dissoluzione della CO2 presente nella fase gassosa e l'idrolisi dell'acido carbonico.
 Qesto sistema chimico ci permette di capire fenomeni di dissoluzione e
concrezionamento delle roccie gessose. Per esempio,
la diffusione di CO2 (spostamento della terza reazione verso
destra) comporta un aumento di ione carbonato e quindi uno spostamento
della seconda reazione verso sinistra, cioe` una deposizione di
carbonato di calcio. Quindi la diminuzione di ione calcio in soluzione
favorisce la dissociazione del solfato, cioe` la dissoluzione del gesso.
In altre parole la diffusione di anidride carbonica rende la soluzione
aggressiva nei confronti del gesso
(effetto Piknett).
Qesto sistema chimico ci permette di capire fenomeni di dissoluzione e
concrezionamento delle roccie gessose. Per esempio,
la diffusione di CO2 (spostamento della terza reazione verso
destra) comporta un aumento di ione carbonato e quindi uno spostamento
della seconda reazione verso sinistra, cioe` una deposizione di
carbonato di calcio. Quindi la diminuzione di ione calcio in soluzione
favorisce la dissociazione del solfato, cioe` la dissoluzione del gesso.
In altre parole la diffusione di anidride carbonica rende la soluzione
aggressiva nei confronti del gesso
(effetto Piknett).
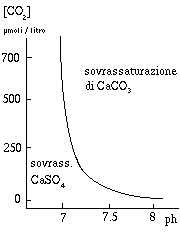
L'analisi del sistema chimico fornisce curve di equilibrio in funzione del pH della soluzione e della concentrazione della CO2 disciolta. La curva varia a seconda della temperatura, in figura e` riportata quella per 10oC. La precipitazione di un qualsiasi sale rende la soluzione sottosatura rispetto all'altro. La precipitazione del gesso innalza il pH, quella della calcite lo abbassa.
E` possibile riscontrare concrezioni calcitiche in grotte di gesso, come pure concrezioni gessose in rocce calcaree. La presenza di carbonati nelle rocce gessose e` dovuta a coperture, intercalazioni, riepimenti, e anche all'azione della CO2 stessa. La presenza di solfati in roccie calcaree e` dovuto a fenomeni indiretti: ossidazione di solfuri da parte di batteri, solubilizzazione di gesso o anidrite.
Il sale (halite) ha una solubilita` molto alta, 360 gr/l. La costante di equilibrio della dissoluzione di NaCl e` K=(Na+)(Cl-) = 101.51 (a 0oC, ma varia poco con la temperatura). La cinetica e` percio` controllata dalla diffusione. Nelle pozze di acqua ferma si arriva all'equilibrio in alcune ore. Nei veli sulle pareti in flusso laminare ci voogliono alcuni minuti. I torrenti turbolenti restano sottosaturi per parecchie centinaia di metri.
Il maggior peso dell'acqua salata, rispetto a quella pura, provoca fenomeni di stratificazione nell'acquifero.
La dissoluzione dle quarzo e` un processo di idratazione,
La costante di equilibrio vale K = (H4SiO4) = 1.1 10-4 (a 25oC). L'acido silicico si deidrata parzialmente formando polimeri e si dissocia liberando ioni H+. La concentrazione di equilibrio e` circa 10 mgr/l. La cinetica e` controllata dalla rottura dei legami chimici e dalla idratazione dei silicati ed e` molto lenta, a temperatura ambiente (10-17 M cm2/sec; puo` aumentare con la presenza di cationi e composti organici).
La dissoluzione nella quarzite e` limitata alle superfici di separazione fra i cristalli, e procede fino a che la roccie diventa sabbia. L'acqua meteorica penetra tra giunti e superfici interstrato, dissolvendo la quarzite ai lati. I maggiori condotti comunque si formano per erosione meccanica, distaccamento dei grani uno ad uno, da parte dell'acqua. Incominciano alle emergenza e procedono a ritroso nel massiccio. Percio` le gallerie sono vadose.
Il carsismo del ghiaccio piu` che un fenomeno chimico e` un fenomeno fisico, tuttavia lo incodo in questa sezione sui meccanismi di carsismo.
[FIXME ...]