In Cosa consiste?
La Trisomia 18 è dovuta alla presenza di un cromosoma supplementare 18. (tre cromosomi 18 anzicchè 2)
Si presenta in circa 1 in 6.000 neonati. Più di 90% dei nati con Trisomia 18 è completo, mentre il resto avrà un mosaicismo (due linee differenti delle cellule, una sana ed una affetta da trisomia).
Come si Manifesta
I neonati affetti da trisomia 18 hanno solitamente ritardo di crescita alla nascita.
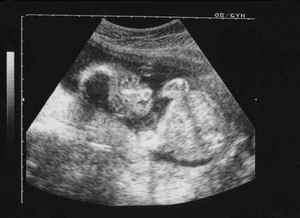
Carattersitiche tipiche della trisomia 18 sono:La protuberanza alla parte posteriore della testa, segno ecografico del cranio a limone, taglio corto delle palpebre, bocca e mascella piccole (micrognazia), variazioni di forma dell'orecchio, del pugno serrato segno del "clenched fist" con sovrapposizione sul 3rd dito da parte del secondo, 4th e 5th. di piccole unghie, dei pollici sottosviluppati o alterati, dello sterno corto (sterno), dei piedi a forma di randello con dorsiflessione dell'alluce, pelle ridondante nella parte posteriore del collo (Aumento della plica nucale) .
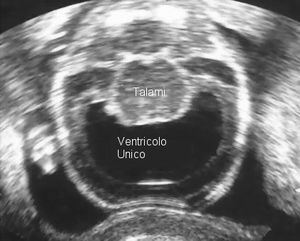
Più di 90% dei bambini con trisomia 18 avranno una malformazione congenita del cuore; tra cui: difetto septale ventricolare - difetto septale atriale - difetti arteriosi di duttali - In più, i bambini con trisomia 18 hanno solitamente un'alterazione di una delle quattro valvole del cuore. Questa combinazione si riferisce a come difetto septale ventricolare con displasia polivalvolare. Circa 10% dei bambini con trisomia 18 avrà un difetto grave del cuore Come ventricolo destro a doppia uscita o il cuore di sinistra ipoplasico.
Diagnosi
Si avvale delle varie Tecniche di diagnostica invasiva delle cromosomopatie: Ecografia Amniocentesi , Villocentesi, Amniocentesi rapida, Villocentesi rapida QF-PCR.
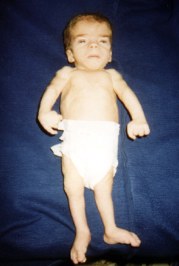
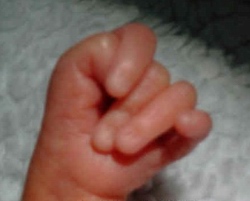
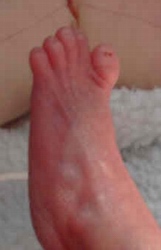
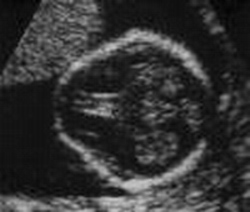
Segno ecografico del Cranio a Limone