Problemi dell'apparato sessuale nella specie umana
generalitŕ
L'apparato sessuale č un apparato estremamente complesso che si forma a partire da diversi tessuti
a livello embrionale.
In termini tecnici possiamo differenziare i problemi che possono colpire l'apparato sessuale in:
1) malattie a trasmissione sessuale (cause esterne)
2) cromosomopatie (causa genetica)
3) altre patologie (cause interne di prevalente origine genetica)
4) disfunzioni sessuali (cause sia interne che esterne)
1) malattie a trasmissione sessuale
Cosa c'č di piů eccitante di una nuova avventura sentimentale, di una nuova conquista, di un incontro magico che culminano subito in un coinvolgente e passionale atto sessuale? Nulla, probabilmente.
Tuttavia, la magia di questi momenti deve fare i conti con una serie di pericoli e rischi che purtroppo vanno considerati ai fini dell'applicazione di una serie di misure preventive che proteggano da essi.
Stiamo parlando della possibilitŕ che questi sconosciuti quanto intriganti compagni d'avventura ci lascino oltre il ricordo di un momento passionale, anche malattie che hanno come loro naturale mezzo di trasmissione l'atto sessuale.
Le Malattie Trasmesse Sessualmente (MTS, STD in inglese) fanno parte delle malattie infettive. Si tratta di oltre venti malattie causate da batteri, virus, funghi o parassiti. In genere si trasmettono tutte per via sessuale, ma si differenziano per la contagiositŕ, il decorso della malattia e le possibilitŕ di cura e prevenzione.
Le malattie veneree "classiche" (per es. la sifilide e la gonorrea) non incutono piů tanta paura da quando sono comparsi gli antibiotici.
Contro l'epatite B, una grave malattia trasmessa anch'essa sessualmente, esiste da tempo un vaccino.
La MTS piů temibile č oggi certamente l'Aids poiché non č stato ancora messo a punto un farmaco in grado di vincere la malattia e non esiste ancora un vaccino.
Ogni anno in tutto il mondo 330 milioni di persone contraggono una malattia a trasmissione sessuale. La piů diffusa č la tricomoniasi (120 milioni di casi), seguono le infezioni da clamidia (50 milioni) e la gonorrea (25 milioni). Il numero dei sieropositivi č stimato attualmente a 33,6 milioni.
Il contagio avviene generalmente durante i rapporti sessuali attraverso il contatto diretto di liquidi organici infetti (sperma e secreti vaginali) con le mucose.
Gran parte delle malattie veneree, in particolare le infezioni da herpes, sono trasmissibili anche tramite rapporti orali, baci e "petting" (contatto diretto con i liquidi organici infetti).
Il contagio puň anche prodursi tra una madre sieropositiva e il feto o il neonato. Alcune malattie veneree (epatite B/C, Aids e sifilide) possono essere trasmesse tramite le conserve di sangue e i prodotti ematici o lo scambio di siringhe tra tossicodipendenti. Alcune infezioni si contraggono tramite l'uso in comune di asciugamani, articoli da toeletta e altro, ma si tratta di casi estremamente rari. Un contagio con goccioline (tosse, starnuti) č escluso. Nei comuni contatti sociali (lavori di casa, ufficio, mezzi pubblici, viaggi e simili) č impossibile infettarsi a condizione che si rispettino le abituali norme igieniche.
La maggior parte delle MST (ad eccezione soprattutto dell'epatite B e dell'Aids) si manifesta inizialmente nelle zone del corpo in cui č avvenuto il contatto, quindi sul pene, nella vagina e sulle labbra della vulva. Possono essere colpiti anche l'ano e la cavitŕ orale. Alcune di queste malattie sono semplicemente fastidiose, altre invece molto pericolose: se non curate possono estendersi a tutto il corpo e causare danni in parte gravi e irreparabili, come sterilitŕ, lesioni cerebrali o cecitŕ
Condilomi acuminati
Detti anche "creste di gallo", sono delle piccole escrescenze di colore rosso, rosato, o bianco che attaccano i genitali, provocate dal cd. Papilloma Umano. I virus del Papilloma sono trasmessi sessualmente per contatto con persone infette e possono causare verruche nella zona genitale ed anale sia degli uomini che delle donne. Tuttavia circa il 50% degli individui infetti non sviluppano mai verruche genitali, ma sono comunque capaci di trasmetter il virus agli altri.
Fino a qualche tempo fa l'unica soluzione era l'intervento chirurgico, ora invece possono essere sufficienti semplici pomate. Alcun specie del virus papilloma sono associate ad aumento del tumore alla cervice. Inoltre le lesioni possono favorire il diffondersi di altre malattie a trasmissione sessuale quali l'Aids. L'uso dei profilattici di lattice o di poliuretano durante i rapporti sessuali puň aiutare a ridurre il rischio di trasmissione, ma č bene sottolineare che la trasmissione puň comunque avvenire se le verruche sono in parti del corpo non coperte dal profilattico. E quindi altro accorgimento č di limitare il numero dei partner di sesso.
Vaginiti
Si presentano con un aumento delle secrezioni vaginali, maleodoranti. Le vaginiti batteriche croniche provocano infiammazione delle tube con conseguente possibile infertilitŕ, gravidanze extrauterine o parto prematuro. Nella metŕ dei casi esse non danno sintomi e possono esser curate con terapia antibiotica.
Clamidia
L'infezione da Clamidia č molto comune tra gli adulti giovani e tra gli adolescenti. Tuttavia molte persone non sanno di avere questa malattia poiché, sebbene siano infettate, non hanno nessun sintomo; di fatti circa il 75% delle donne infette e metŕ degli uomini infetti non presentano sintomi di Clamidia. La trasmissione avviene attraverso contatto sessuale (soprattutto vaginale o anale) con una persona infetta. Nelle donne i sintomi della Clamidia possono includere l'aumento delle secrezioni vaginali, bruciori, emorragie dopo i rapporti sessuali; nell'uomo sintomi possono essere bruciori e testicoli gonfi e dolorosi. La cura č generalmente a base di antibiotici. Se trascurata essa puň provocare infertilitŕ in uomini e donne e in queste ultime gravidanze extrauterine.
Herpes genitale
Pochi giorni aver contratto questo virus, nell'area genitale compaiono lacerazioni che rimangono per due o tre settimane. Il virus in seguito si rifugia all'interno di alcune cellule nervose del midollo spinale, dove resta inattivo per poi ripresentarsi periodicamente. Coloro che lo hanno contratto possono esser contagiosi anche in assenza di lesioni visibili e per causare il contagio č sufficiente anche un contato con le zone vicine ai genitali. L' herpes si tiene sotto controllo con farmaci antivirali e la terapia puň durare anche alcuni anni.
Tricomoniasi
Il Trichomonas vaginalis č un organismo microscopico che causa la malattia Tricomoniasi, che puň essere trasmessa sessualmente da persona a persona. La Tricomoniasi č trasmessa attraverso rapporti di sesso vaginale con una persona infetta. I fattori di rischio primari della Tricomoniasi includono l'intraprendere sesso con una persona della quale non si conoscono le abitudini in fatto di igiene e vita sessuale, fare sesso con piů partner, avere una relazione con qualcuno che ha piů partners. Sia gli uomini che le donne possono essere infettate ma spesso non si hanno sintomi dell'infezione per cui non si sa di avere la malattia. Nelle donne il sintomo maggiore č l'aumento delle secrezioni vaginali, con cambio di colore e cattivo odore, bruciori della vagina. Nell'uomo scarico del pene e bruciori durante l'urinazione.
Gonorrea
La Gonorrea č un'infezione causata dai batteri Neisseria gonorrhoeae. La Gonorrea puň portare all'infezione dell'uretra, della cervice, del retto e della gola. Tuttavia molte persone non sanno di avere la Gonorrea, poiché, sebbene siano infettate, non hanno nessun sintomo. La Gonorrea č trasmessa attraverso contatto sessuale (sesso vaginale, anale o orale) con una persona infetta. La Gonorrea puň avere un effetto deleterio sui genitali, sul retto o sulla gola. Molte donne e molti uomini con la Gonorrea non presentano sintomi evidenti, soprattutto con l'infezione del retto e della gola. Nelle donne i maggiori sintomi sono aumento delle secrezioni vaginali, bruciori, piccole emorragie; negli uomini bruciori durante l'urinazione, testicoli doloranti. L'infezione del retto puň accadere facendo sesso anale ricettivo. Nelle donne, l'infezione del retto accade piů frequentemente attraverso diffusione dell'infezione dalla vagina. Sebbene non vi siano spesso sintomi di infezione del retto, essi possono includere disturbi del retto, prurito dell'ano, dolore, scarico o emorragia. L'infezione della gola puň accadere facendo sesso genitale-orale con partner infetti. Nella gola, la Gonorrea puň causare una gola infiammata.
Sifilide
Provocata dal batterio Treponema pallidum, si manifesta dopo tre mesi dal contagio ed č caratterizzata da ulcerazioni nella parte della pelle che č venuta a contatto con quelle del partner malato. Se non curata puň intaccare il sistema nervoso e portare alla morte. Si cura con la penicillina. Il primo sintomo della Sifilide č di solito un piccola ferita indolore (sifiloma) nella zona a contatto sessuale (pene, vagina, ano, retto, o bocca). La ferita di solito appare 2-6 settimane dopo l'esposizione e sparisce entro poche settimane. Brevemente dopo che la ferita guarisce, si puň rilevare un'eruzione cutanea in tutto il corpo (inclusi i palmi delle mani e le piante dei piedi), linfonodi gonfi, febbre o affaticamento. Questi sintomi spariscono entro poche settimane. Anche se i sintomi iniziali della Sifilide svaniscono da soli, il batterio della Sifilide rimarrŕ nel corpo se non viene curato.
Epatite B
L'Epatite B č una malattia seria del fegato che č causata dal virus dell'Epatite B. Essa č estremamente contagiosa
e puň essere trasmessa sessualmente o per contatto con sangue o liquidi del corpo infetti. Sebbene il virus dell'Epatite
B puň infettare le persone di tutte le etŕ, i giovani adulti e gli adolescenti sono quelli a rischio maggiore.
Il virus dell'Epatite B attacca direttamente il fegato e puň portare a malattie intense, danni del fegato, e in alcuni
casi alla morte. Sebbene non esista nessuna cura per l'Epatite B, esiste un vaccino sicuro ed efficace che puň
prevenire la malattia. Il virus dell'Epatite B č altamente contagioso e si diffonde attraverso contatto con il
sangue e con altri liquidi del corpo (inclusi lo sperma, le infezioni vaginali e latte materno) di individui infetti.
Esso puň essere trasmesso attraverso il contatto sessuale (vaginale, anale o orale) con una persona infetta;
scambio di siringhe; uso di lamette da barba o aghi per tatuaggi; gravidanza e/o parto. Sebbene non esiste nessuna
cura per il virus dell'Epatite B, esiste un vaccino sicuro ed efficace che puň prevenire l'Epatite B. Questo vaccino
č stato reso disponibile sin dal 1982 ed č fornito in serie di tre iniezioni. Esso fornisce protezione contro l'Epatite
B in 90-95% di quelli vaccinati. Essere vaccinati č la migliore maniera di ridurre il rischio di contrarre l'Epatite B.
2) cromosomopatie
Come ricordato sono di prevalente origine genetica, derivanti dalla presenza o assenza di pezzi di cromosomi.
- La sindrome di Down o trisomia 21
- La sindrome di Edwards o trisomia 18
- La sindrome di Patau o trisomia 13
- La sindrome di Turner
- La sindrome di Klinefelter
- La sindrome del X Fragile
La sindrome di Down č la piů frequente causa di ritardo mentale. Nota fin dal XVI° secolo ha avuto il suo
inquadramento nosologico solo nel 1866 ad opera di John Longdong Down, l'illustre medico inglese da cui prende il
nome, che ha dedicato tutta la sua vita allo studio e alla cura dei bambini con anomalie psichiche, e che per primo
ha descritto le caratteristiche di un gruppo di bambini con ritardo mentale e tratti orientaleggianti del viso,
definendoli come affetti da "idiozia mongoloide".
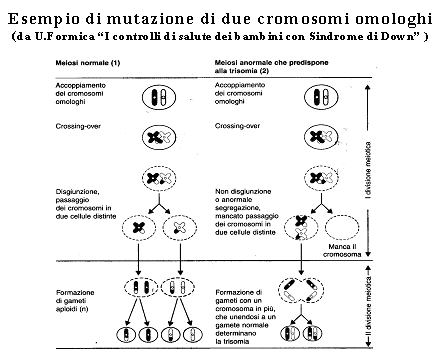
Nel 1959 J.Lejeune e al. hanno dimostrato che la SD dipende dalla presenza di un cromosoma in piů nella coppia 21,
e da allora viene anche definita "Trisomia 21".
Data la casualitŕ della sua comparsa č una anomalia cromosomica congenita non ereditaria, ed č anche la piů nota e
diffusa, perché la lunga sopravvivenza delle persone che ne sono portatrici ha determinato nella societŕ la
consapevolezza della sua presenza.
Come si manifesta: Alla nascita, le caratteristiche fisionomiche dei bambini con SD alla nascita sono principalmente
le seguenti:
- Viso rotondo con sella nasale larga e appiattita
- Occhi con taglio delle palpebre di tipo orientale
- Piega cutanea all'angolo interno degli occhi (epicanto)
- Orecchie piccole
- Collo tozzo con plica nucale abbondante e lassa
- Mignoli inclinati verso l'interno della mano (bradiclinodattilia)
- Solco palmare unico in entrambe le mani
- Marcata ipotonia muscolare
- Articolazioni molto flessibili per l'eccessiva lassitŕ dei legamenti
Ovviamente non č evidente alla nascita il ritardo mentale, che č ben noto a tutti come intrinseco ai trisomici 21
e che rappresenta la caratteristica che piů preoccupa in questa sindrome. Poiché questi tratti fisionomici possono
essere presenti, in modo piů o meno evidente, anche in neonati non trisomici, la diagnosi di certezza di SD si ottiene
solo dallo studio del cariotipo, che si evidenzia eseguendo la mappa cromosomica. L'indagine, indicata anche con il
termine di analisi citogenetica, č la sola che autorizza la diagnosi di Trisomia 21. Consiste nell'analizzare e
fotografare al microscopio, con particolari tecniche, i cromosomi di alcune cellule del sangue (in genere linfociti),
prelevate dal bambino e bloccate in mitosi, e rende possibile il riscontro di una trisomia primaria o da traslocazione.
Pertanto deve essere eseguita subito alla nascita ogni volta che si avanza il sospetto di Trisomia 21 su base clinica,
al fine di avere una certezza diagnostica incontrovertibile.
La sindrome di Edwards (o trisomia 18) appartiene al gruppo delle patologie determinate da alterazioni del
numero o della struttura dei cromosomi. Si stima che la sindrome di Edwards si presenti in 1 neonato su 6000.
Come si manifesta: I neonati affetti presentano grave ritardo di crescita (anche nel periodo prenatale), grave
ritardo mentale, malformazioni multiple: micrognazia (mascella di dimensioni inferiori alla norma), occipite
prominente, alterazioni degli arti, cardiopatia, anomalie renali. Alla sindrome sono associati moltissimi difetti
che causano morte prematura.
Le cause: La sindrome di Edwards č detta anche trisomia 18, perchč č determinata dalla presenza di 3 copie del
cromosoma18, anziché di 2 copie. Il numero totale di cromosomi di un individuo affetto dalla sindrome di Edwards
č quindi 47 e non 46, come negli individui sani. Nella maggior parte dei casi il meccanismo che causa la trisomia
č la "non disgiunzione" dei cromosomi durante la divisione cellulare che avviene nel corso della formazione degli
ovociti: anziché separarsi l'uno dall'altro nelle due cellule figlie, i 2 cromosomi 18 vanno entrambi nella stessa
cellula figlia. Pertanto, in seguito alla fecondazione, con il contributo paterno il cromosoma 18 sarŕ presente in
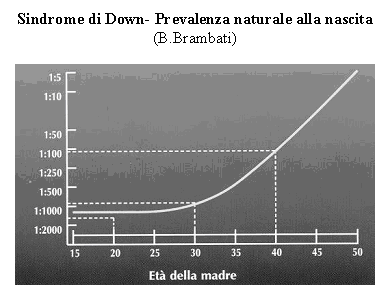
3 copie anziché in 2 copie. E' stato dimostrato che l'etŕ materna č un fattore di rischio per eventi di non disgiunzione:
all'aumentare dell'etŕ tale evento si verifica piů frequentemente.
La diagnosi: La diagnosi eseguita sulla base delle caratteristiche cliniche della sindrome, trova conferma nell'analisi
dei cromosomi (v. scheda sulle anomalie cromosomiche), che permette di evidenziare la trisomia del cromosoma 18.
L'esame del liquido amniotico, dei villi coriali e del sangue fetale permettono di analizzare i cromosomi del feto
e quindi di evidenziare la presenza di eventuali anomalie cromosomiche, compresa la sindrome di Edwards.
Inoltre l'esame ecografico dei feti con trisomia 18 mostra della anomalie nella quasi totalitŕ dei casi.
Tali anomalie consistono in ritardo di crescita e in varie malformazioni congenite quali, ad esempio, omfalocele,
aplasia del radio e igroma.
La sindrome di Patau o trisomia 13 indica la presenza di tre copie del cromosoma 13 invece di due, per un
totale di 47 cromosomi invece di 46.
La condizione clinica che viene causata da questo squilibrio cromosomico, come ad esempio: scarso peso alla nascita
uguale o inferiore al 10 percentile, malformazioni degli organi interni variabilmente gravi e notevole ritardo dello
sviluppo.
Incidenza La Trisomia 13 č una malattia rara, ad elevata mortalitŕ, ha un incidenza annuale di un bambino su 10000.
In ogni caso, la Trisomia 13 rimane la causa cromosomica di numerosi casi di morte del feto, aborto e nascita di feto
morto.
Sopravvivenza La sopravvivenza č scarsa, varia a seconda del sesso, ed č piů favorevole per le femmine.
Ad un anno di etŕ il 38% degli individui era vivente, a 5 anni il 13%, a 10 anni il 3%. Il piů vecchio soggetto al
momento della ricerca aveva 9 anni ed č vissuto fino ad 11.
La media di vita č stata di 12,8 mesi: nei maschi 3,6 mesi, nelle femmine 20,4 mesi.
Malformazioni Associate:
- Malformazioni cardiache 64%
- Labbro leporino 39%
- Palatoschisi 42%
- Piede equino 9%
- Ompalocele 6%
- Polidattilia 67%
- Difetti al cuoio capelluto 42%
- Difetti agli occhi 64%
- Vista rovinata 46%
- Attacchi epilettici 42%
- Problemi renali 6%
- Infezioni urinarie 21%
- Spina bifida 3%
- Difetti cerebrali 18%
In aggiunta a questi difetti strutturali e complicanze mediche, sono segnalate anche contratture, genitali anormali,
lussazioni, scoliosi e sindattilia.
Cause di morte La piů comune causa di morte č stata l'arresto Cardiopolmonare (69%), seguita dalle patologie congenite
al cuore (13%) e da cause polmonari (4%).
Diagnosi Si avvale delle varie Tecniche di diagnostica invasiva delle cromosomopatie: Amniocentesi , Villocentesi,
QF-PCR.
La sindrome di Turner č una condizione caratterizzata dalla presenza di un unico cromosoma X nei soggetti di
sesso femminile (normalmente caratterizzati da due cromosomi sessuali XX) che, pertanto, vengono indicati come X0.
La sindrome di Turner ha una frequenza di circa un caso su 2000-2500 bambine nate vive; fu descritta nel 1938 dal medico
Harry Turner. Una elevata percentuale di feti portatori dell'assetto cromosomico XO vengono abortiti spontaneamente.
Cause La formazione di uno zigote con un solo cromosoma sessuale X, da cui si sviluppa un individuo portatore di
sindrome di Turner, avviene se una cellula uovo normale, con corredo cromosomico X, viene fecondata da uno
spermatozoo 0, ovvero privo di cromosoma sessuale; oppure se uno spermatozoo portatore del cromosoma X si fonde con una
cellula uovo priva di cromosoma sessuale. Le anomalie dei gameti sono causate da errori nel corso della meiosi, cioč
durante il processo di formazione dei gameti. In particolare, la mancata disgiunzione della coppia di cromosomi sessuali
puň determinare la formazione di spermatozoi XY e 0; oppure di cellule uovo XX e 0.
Come si Manifesta La sindrome di Turner comprende un ampio spettro di caratteristiche che non si presentano
necessariamente insieme; anche la gravitŕ delle patologie non č la stessa per tutti i soggetti. I soggetti XO si
distinguono in genere per la bassa statura (altezza media: 1,45 m) e ritardi nel processo di ossificazione dello
scheletro; possiedono una gabbia toracica di forma anomala, piuttosto ampia e piatta (torace "a scudo"); vanno
soggetti a osteoporosi piů precocemente e in modo piů grave rispetto alle donne XX. In questi individui, alcune ossa
metacarpali (cioč le ossa localizzate nel palmo della mano), in particolare la quarta e la quinta, sono piů corte del
normale; in circa il 10% dei casi, sono presenti anche anomalie dell'apparato circolatorio, che nella maggior parte
dei casi consistono nel restringimento di alcune arterie. Possono manifestarsi anche, in circa un terzo dei casi,
anomalie nell'anatomia dei reni (rene con una forma "a zampa di cavallo" o bacinetto renale biforcato in due estremitŕ)
e delle ovaie. Circa nella metŕ dei casi, durante lo sviluppo embrionale dei soggetti XO si sviluppa l'igroma cistico,
patologia che comporta la formazione di una o due grosse cisti piene di linfa nella regione posteriore del cranio;
queste strutture disturbano la comunicazione tra il sistema linfatico e le vene. Le donne con sindrome di Turner in
genere non hanno ciclo mestruale e sono sterili. La diagnosi della sindrome puň avvenire mediante diagnosi prenatale,
attraverso amniocentesi o esame dei villi coriali, o, al momento della nascita, dalla valutazione delle caratteristiche
fisiche della neonata. In alcuni casi, la condizione X0 puň diventare evidente solo nel corso dell'infanzia, durante
la quale lo sviluppo appare piů lento rispetto a quello dei coetanei.
Terapia: poiché la condizione XO si produce al momento della formazione dello zigote, e fa parte del corredo cromosomico
dell'individuo, non vi sono cure che possano modificarla. E' possibile perň intervenire in modo da ovviare ad alcune
carenze associate alla sindrome di Turner. La somministrazione di ormone della crescita (Somatotropina), ed eventualmente
di ormoni androgeni, puň stimolare lo sviluppo corporeo. Le anomalie ovariche comportano una carente produzione di ormoni
estrogeni, il che comporta disturbi nello sviluppo dei caratteri secondari femminili; una terapia ormonale dall'etŕ di
circa 12 anni puň ovviare a tale carenza e permettere il raggiungimento di caratteristiche normali.
L'infertilitŕ delle
donne X0 puň essere superata con tecniche di riproduzione assistita. Le patologie cardiovascolari e renali associate
alla sindrome di Turner, nei soggetti che ne soffrono, rappresentano l'aspetto piů delicato per la salute del paziente
Turner, poiché possono dare luogo a complicazioni; pertanto, richiedono un costante controllo medico.
La sindrome di Klinefelter appartiene al gruppo delle anomalie cromosomiche patologie determinate da alterazioni
del numero o della struttura dei cromosomi.
Si stima che la sindrome di Klinefelter presenti una frequenza alla nascita di 1 su 1000 neonati maschi
Come si manifesta: Caratteristiche generali sono: ipogonadismo (testicoli di dimensioni ridotte), bassi livelli di
testosterone, mancata produzione di spermatozoi (azoospermia) e quindi sterilitŕ; possono essere presenti una
sproporzione tra lunghezza degli arti e lunghezza del tronco e statura superiore alla media. Spesso la diagnosi viene
posta solo in etŕ adulta, nel corso di analisi eseguite per infertilitŕ di coppia: č stato evidenziato che il 10% dei
maschi azoospermici (con liquido seminale privo di spermatozoi) presenta sindrome di Klinefelter.
Le cause: I soggetti affetti da questa sindrome hanno 47 cromosomi, con un cromosoma X sovrannumerario (cariotipo 47,
XXY). La sindrome di Klinefelter puň presentarsi anche in mosaico: in questo caso i pazienti presentano sia cellule
con cariotipo 46, XY sia cellule con cariotipo 47, XXY; in questi pazienti le caratteristiche cliniche della sindrome
sono attenuate.
La diagnosi: La diagnosi oltre che sulle caratteristiche cliniche della sindrome si basa anche sull'analisi dei
cromosomi, che permette di evidenziare l'alterazione del numero dei cromosomi (v. scheda sulle anomalie cromosomiche),
confermando cosě la diagnosi. L'esame del liquido amniotico, dei villi coriali e del sangue fetale permettono di
analizzare i cromosomi del feto e quindi di evidenziare la presenza di eventuali anomalie cromosomiche, compresa
la sindrome di Klinefelter (v. scheda sulla diagnosi prenatale).
Altre informazioni sulla sindrome di Klinefelter: il termine sindrome di Klinefelter (KS) descrive un gruppo di
patologie cromosomiche in cui č presente almeno un cromosoma X in soprannumero, rispetto al cariotipo normale
maschile, 46,XY. L'aneuploidia XXY č l'anomalia dei cromosomi sessuali piů frequente nei maschi, con una frequenza
di 1/500. Le varianti della sindrome di Klinefelter sono molto piů rare e il cariotipo 48,XXYY e 48,XXXY č presente
in circa 1/50.000 neonati maschi. La prevalenza di 49,XXXXY č 1/85.000-100.000 neonati maschi e rappresenta la variante
piů grave di sindrome di Klinefelter. L'analisi citogenetica classica č necessaria per formulare la diagnosi definitiva;
i segni clinici piů gravi sono presenti nei maschi portatori di un numero progressivamente crescente di cromosomi
sessuali. Per la diagnosi viene utilizzata l'analisi cromosomica sui linfociti del sangue periferico, o sugli amniociti
o sui villi coriali, nel caso di diagnosi prenatale. Quando la diagnosi non viene posta nel
periodo prenatale, i maschi 47,XXY possono presentare una varietŕ di segni clinici sfumati che correlano con l'etŕ.
Nell'infanzia, i maschi 47,XXY possono essere indirizzati all'analisi cromosomica per la presenza di ipospadia,
pene piccolo o criptorchidismo. Il bambino in etŕ scolare puň presentare ritardo del linguaggio, difficoltŕ
nell'apprendimento o problemi comportamentali. Il bambino piů grande o l'adolescente possono essere diagnosticati
nel corso di una valutazione endocrinologica, per il ritardo o l'incompletezza dello sviluppo puberale, che si
associano ad un habitus eunucoide, ginecomastia e testicoli di volume ridotto. Gli adulti sono spesso valutati per
infertilitŕ o tumori maligni del seno. La terapia sostitutiva a base di androgeni dovrebbe iniziare durante la pubertŕ,
intorno all'etŕ di 12 anni, con dosi crescenti sufficienti a mantenere concentrazioni sieriche di testosterone,
estradiolo, FSH e LH appropriate all'etŕ. La terapia sostitutiva č utile a normalizzare le proporzioni del corpo
e ad incidere sullo sviluppo delle caratteristiche sessuali secondarie; inoltre previene lo sviluppo della ginecomastia.
Il cromosoma X in soprannumero nella sindrome di Klinefelter origina sporadicamente sia dalla non-disgiunzione meiotica,
per mancata separazione di un cromosoma durante la prima o la seconda divisione nel corso della gametogenesi, sia dalla
non-disgiunzione mitotica, nello zigote in fase di sviluppo. La probabilitŕ di non-disgiunzione del cromosoma X aumenta
con l'aumento dell'etŕ materna. Gli effetti sullo sviluppo fisico e mentale aumentano con l'aumento del numero degli X
in soprannumero; ogni X soprannumerario riduce il QI complessivo di 15-16 punti, con un coinvolgimento maggiore del
linguaggio, in particolare delle capacitŕ espressive.
La sindrome da X fragile (FraX) č la forma piů comune di ritardo mentale dopo la sindrome di Down e la piů
frequente fra quelle ereditarie. La malattia č causata dall'alterazione di un gene chiamato FMR1 (Fragile Mental
Retardation 1), situato nel cromosoma X. Questa alterazione nella maggior parte dei casi porta all'assenza o alla
carenza della proteina FMRP codificata da questo gene.
La funzione di FMRP non č ancora nota con certezza, ma la proteina sembra essere implicata nei processi neuronali
che stanno alla base dell'apprendimento e della memoria. Il nome "X fragile" deriva dal fatto che l'alterazione del
gene FMR1 provoca delle modificazioni nella struttura del cromosoma X, che al microscopio presenta una strozzatura
in un punto preciso (quello in cui č situato il gene).
Come si manifesta: La sindrome del FraX colpisce molto piů frequentemente i maschi rispetto alle femmine, dato che
queste ultime possiedono 2 copie del cromosoma X. Lo sviluppo mentale delle persone affette da FraX č molto vario:
alcune mostrano capacitŕ cognitive quasi normali, altre un lieve ritardo mentale, altre ancora un ritardo mentale
piů grave. I sintomi di seguito descritti possono presentarsi in forma variabile:
ritardo cognitivo Il primo segno della malattia č il ritardo nello sviluppo psicomotorio, in particolare
nell'apprendimento del linguaggio. Il ritardo mentale č di grado variabile e spesso si associa ad anomalie
comportamentali come irrequietezza, instabilitŕ psicomotoria e incapacitŕ a fissare l'attenzione.
Queste caratteristiche persistono con l'avanzare dell'etŕ.
Anomalie nel comportamento Il comportamento delle persone affette da FraX puň andare da un carattere estroverso
e sociale a comportamenti simili all'autismo (iperattivitŕ, incapacitŕ di fissare negli occhi gli altri, avversione
all'essere toccati, comportamento stereotipato). A volte possono manifestarsi anche episodi convulsivi.
Caratteristiche fisiche: molte persone affette da FraX hanno tratti somatici tipici: viso stretto e allungato con
fronte e mandibola prominenti, orecchie piů grandi e piů basse della media e ingrossamento dei testicoli (macrorchidismo). Le persone affette da FraX possono presentare anche altri sintomi, come l'iperestensibilitŕ delle articolazioni, il piede piatto e il prolasso della valvola mitrale (un'anomalia di una valvola cardiaca).
Le cause: Nel 1991 č stato identificato il gene FMR1 in corrispondenza del sito fragile del cromosoma X.
Il gene FMR1 codifica una proteina, chiamata FMRP (Fragile X Mental Retardation Protein), di cui non č ancora chiara
la funzione. Nella maggior parte dei casi di FraX l'alterazione responsabile della sindrome consiste nell'espansione,
attraverso le generazioni, di un tratto di DNA di questo gene, composto da tre basi nucleotidiche ripetute (CGG).
Mentre nelle persone normali queste basi sono ripetute in un numero variabile da 6 a 55 volte, nelle persone malate
sono ripetute piů di 200 volte. Questa espansione (indicata come "mutazione completa"), insieme ad altri fenomeni,
provoca il mancato funzionamento del gene FMR1 e la conseguente assenza della proteina da esso codificata. Quasi tutti
i maschi con la mutazione completa presentano i sintomi della malattia, mentre circa la metŕ delle femmine presentano
ritardo mentale di grado variabile (da lieve a grave). Ci sono inoltre alcuni casi rari di FraX in cui l'alterazione
responsabile della sindrome non consiste nell'espansione ma č dovuta a mutazioni nella regione codificante del gene
e quindi nella proteina. Alcune persone possiedono all'interno del gene FMR1 un numero di ripetizioni intermedie (da
56 a 200), che non provocano alcun effetto. Questa alterazione intermedia č detta "premutazione" e gli individui che
la possiedono sono portatori sani, perché nelle generazioni successive le ripetizioni possono aumentare causando la
mutazione completa. Recentemente sono stati descritti alcuni casi in cui anche portatori di una premutazione presentano
la sindrome. Sebbene non si conosca ancora con certezza la funzione della proteina FMRP, č noto che questa proteina
č presente ad alti livelli nelle cellule neuronali ed č localizzata anche nelle sinapsi, i punti di contatto tra le
cellule nervose a livello dei quali avviene la trasmissione degli impulsi nervosi. In queste sedi sembra che FMRP
sia in grado di modulare l'espressione di alcune proteine implicate nella formazione e nel funzionamento delle sinapsi.
La deregolazione di queste proteine quando non č presente FMRP potrebbe essere la causa del ritardo mentale osservato
nei pazienti affetti dalla sindrome.
Come si trasmette: La modalitŕ di trasmissione presenta alcune caratteristiche peculiari. Maschi e femmine che portano
la premutazione non mostrano alcun sintomo. I maschi che portano la premutazione la trasmettono a tutte le loro figlie
senza che avvengano variazioni (a nessuno dei figli maschi verrŕ trasmessa la premutazione). Le figlie quindi saranno
portatrici della premutazione senza avere alcun sintomo, ma rischiano di avere figli malati, perché durante la
formazione dell'ovulo materno puň avvenire l'espansione delle ripetizioni nel gene FMR1. Quasi tutti i maschi con
la mutazione completa presentano i sintomi della malattia, mentre circa la metŕ delle femmine presentano ritardo mentale
di grado variabile (da lieve a grave). Č possibile individuare sia la premutazione sia la mutazione completa attraverso
indagini di genetica molecolare (esame del DNA), che permettono un'identificazione accurata dei soggetti affetti e dei
portatori. Grazie a questa analisi molecolare č quindi possibile individuare le famiglie a rischio e lo specialista potrŕ
offrire loro un'adeguata consulenza genetica. L'indagine prenatale, eseguibile fin dal primo trimestre di gravidanza,
permette di stabilire se il feto ha ereditato l'alterazione genetica. Le coppie che temono di poter trasmettere la
malattia ai propri figli possono rivolgersi ad un consulente genetico, che potrŕ informarli piů precisamente sulle
possibilitŕ di dare alla luce figli malati, ed eventualmente indicare gli esami appropriati. Come per tutte le malattie
genetiche, č bene fare questo prima di decidere di avere un figlio, perché alcune analisi possono richiedere molto tempo
prima di dare una risposta certa. In ogni caso, č sempre possibile rivolgersi al consulente genetico, anche se la
gravidanza č giŕ iniziata, per una diagnosi prenatale.
La diagnosi: purtroppo, molti pazienti con ritardo mentale o psicofisico sono affetti da FraX senza che questa malattia
sia correttamente diagnosticata. Con il progredire delle conoscenze e delle tecniche diagnostiche, si spera che sempre
piů persone affette da FraX possano ricevere una diagnosi corretta perché, anche se per ora non esiste una terapia
specifica per l'X fragile, la diagnosi č utile per stabilire se i genitori corrano il rischio di avere altri figli
affetti. Fino all'inizio degli anni 90 la diagnosi si č basata su tecniche citogenetiche, cioč sulla ricerca del sito
fragile sul cromosoma X al microscopio con particolari tecniche. Attualmente questa indagine č stata sostituita da un
esame piů sensibile, quello molecolare (esame diretto del DNA), che permette di mettere in evidenza direttamente a
livello del gene FMR1 l'espansione del tratto di DNA ripetuto CGG. Poiché esistono casi di FraX in cui l'assenza
della proteina FMRP non č associata all'amplificazione della tripletta CGG ma ad altri difetti nel gene FMR1 (per
esempio a delezioni genetiche cioč alla mancanza di parti del gene), nel 1999 č stata sviluppata una nuova tecnica
diagnostica di tipo immunocitochimico, che si basa sulla diretta individuazione della proteina FMRP nei linfociti di
uno striscio di sangue o nella radice dei capelli. Questa tecnica non invasiva pur essendo di estrema rapiditŕ, non č
in grado di individuare i portatori di una premutazione o i pazienti che sintetizzano normali quantitŕ della proteina
FMRP mutata. Data l'elevata frequenza della sindrome, si puň sospettarne la presenza ogni qualvolta un soggetto presenti
ritardo psicomotorio e del linguaggio, in particolare se esistono piů individui affetti nell'ambito familiare.
Non esiste attualmente un trattamento specifico per questa sindrome. La terapia rimane quindi di tipo riabilitativo,
sia motorio sia psicopedagogico. Una buona assistenza psicopedagogica da parte di educatori specializzati puň migliorare
sensibilmente le potenzialitŕ del bambino e aiutarlo a vivere i rapporti con gli altri in modo piů armonico.
Patologie associate (quali epilessia, prolasso della valvola mitrale o altre) dovranno essere affrontate dal medico
con adeguate terapie.
3) altre patologie
Poiché le patologie a carico dell'apparato sessuale sono numerose e legate strettametne alla disfunzioni sessuali, se ne scrive
solo un lungo elengo analizzandone solo alcune.
Patologia del pene:
- Ipospadia;
- Recurvatum penino;
- Epispadia;
- Micropene;
- Pene palmato (Webbed penis);
- Pene nascosto (Concealed penis);
- Fimosi.
Patologia del testicolo:
- Criptorchidismo:
- Testicolo non disceso/Ectopia testicolare,
- Testicolo non palpabile,
- Atrofia testicolare primitiva (Vanishing testis);
- Torsione del funicolo spermatico;
- Atrofia testicolare secondaria;
- Orchite;
- Neoplasie benigne;
- Neoplasie maligne
Tra queste patologie analizziamo quelle collegate alla ritenzione dei testicoli. Ogni anomalia che alla nascita
non permette la presenza di entrambi i testicoli nello scroto va intesa come testicolo ritenuto. Si preferisce definire
criptorchidismo solo le condizioni in cui il testicolo non si reperisce né clinicamente né ecotomograficamente (come
l'etimologia greca impone).
Sicuramente la complessitŕ del fenomeno suggerisce una multifattorialitŕ di eventi quale causa del testicolo ritenuto.
Esistono poi molte sindromi in cui il testicolo ritenuto fa parte del quadro sindromico complessivo. E' pertanto bene
tener sempre presente questa ipotesi.
Il testicolo retrattile č quella condizione in cui il testicolo per una retrazione transitoria rimane fuori dallo scroto
per contrazione del muscolo cremasterico. Questo muscolo nell'uomo ha due importanti compiti : quello di regolare la
temperatura e quello di proteggere il testicolo dai traumi. La stimolazione del riflesso avviene a basse temperature
(visitare sempre il bambino in ambulatori caldi) e per stimolazione del lato interno della coscia. Alla nascita questo
riflesso č debole o assente. L'entitŕ del riflesso č notevolmente evidente fino al decimo anno circa per poi diminuire
con l'aumentare dei livelli ormonici maschili. Particolare attenzione va posta al testicolo retrattile nei casi " border
line " per precisare quanto vi sia di fisiologico o di patologia minima.
In numerosi studi l'incidenza del testicolo ritenuto vero si aggira sul 5% della popolazione sana . Bisogna ricordare
sempre che una discesa puň avvenire spontaneamente nei primi tre /sei mesi d'etŕ e che quindi una valutazione va
ragionevolmente posta dopo tale periodo anche dilatabile oltre non essendovi problemi terapeutici urgenti fatte salve
le patologie associate (ernia, torsione ecc.). Nel bambino prematuro, per ovvie ragione di sfalsamento dei tempi di
sviluppo fisiologico ed anatomico, l'incidenza aumenta sino a toccare punte del 60/70% nei pazienti con peso inferiore
ai 1500 grammi.
Il problema centrale č che il testicolo sia nella borsa scrotale , deputata a tenere delle basse temperature con i vari
meccanismi di cui č dotata : il plesso pampiforme,la pigmentazione scrotale ,l'assenza di grasso sottocutaneo e la
regolazione attraverso muscoli sensibili alle variazioni termiche come il muscolo cremastere ed il dartos.
Nell'uomo la temperatura a livello scrotale č di 33°, di 35° nella regione inguinale e di 37° in addome.
Tutto il problema č imperniato sul fatto che un testicolo a temperature piů alte di quelle necessarie e garantite
dalla sede scotale subisce delle alterazioni lievi a partire dal sesto mese e gravi a partire da quarto anno di etŕ.
Tutta la strategia terapeutica va dunque impostata su questo problema e cioč sul fatto che dopo il quarto anno di
etŕ i testicoli debbono essere obbligatoriamente in sede scrotale.
Le tecniche chirurgiche sono ormai perfettamente codificate e prevedono sostanzialmente due modelli operativi.
Il primo č l'intervento chirurgico classico , in un tempo o due tempi a seconda della lunghezza dei vasi spermatici
e del deferente , con una piccola incisione a livello inguinale ed un'altra a livello scrotale che č indicata in tutti
i casi di testicolo ritenuto palpabile (85% dei casi). L'intervento prevede la preparazione dei vasi venosi ed arteriosi
del funicolo e del dotto deferente.
Successivamente viene isolato, legato e sezionato il dotto peritoneo vaginale presente in circa il 70% dei casi
e si attua di fatto l'erniotomia.
Il testicolo viene quindi posizionato e fissato in una tasca extradartotica scrotale.
Il secondo č l'approccio video-laparoscopico in due tempi limitatamente ai casi di testicolo non palpabile (15% dei casi).
Le complicanze sono relativamente rare e si riferiscono prevalentemente alla sepsi ed alle conseguenze della lesione
incidentale dei vasi spermatici o del deferente con atrofia secondaria del testicolo.
I risultati sono sostanzialmente buoni sia per il posizionamento del testicolo che per la fertilitŕ a distanza anche se l'interpretazione dei risultati rimane non semplice per i tempi di valutazione nell'arco di circa un ventennio con diversi approcci chirurgici difficilmente comparabili. Sicuramente un'ampia revisione della letteratura dimostra un aumento della fertilitŕ per l'intervento precoce soprattutto per quei testicoli posti in prossimitŕ del canale inguinale.
Patologia dell'epididimo:
- Cisti;
- Epididimiti.
Patologia del canale inguinale:
- Ernia inguinale;
- Idrocele;
Cisti del funicolo spermatico.
Varicocele
Patologia ginecologica:
- Malformazioni uterine;
- Malformazioni vaginali:
- intersessi,
- agenesia vaginale,
- setti vaginali: imene imperforato e setti vaginali trasversi - longitudinali;
- Sinechie piccole labbra;
- Corpi estranei;
- Polipi imenali;
- Lichen sclero-atrofico;
- Prolasso uretrale;
- Cisti parauretrali;
- Emangiomi;
- Ovaio policistico;
- Tumori.
Traumi:
- Lesione accidentale;
- Abusi sessuali.
4) disfunzioni sessuali
Una Disfunzione Sessuale č caratterizzata da un'anomalia del processo che sottende il ciclo di risposta sessuale, o da dolore associato al rapporto sessuale. Il ciclo di risposta sessuale normale puň essere diviso nelle seguenti fasi:
- Desiderio. Questa fase consiste in fantasie sull'attivitŕ sessuale e nel desiderio di praticare attivitŕ sessuale.
- Eccitazione. Questa fase consiste in una sensazione soggettiva di piacere sessuale e nelle concomitanti modificazioni fisiologiche. Le principali modificazioni nel maschio sono la tumescenza del pene e l'erezione. Le principali modificazioni nella donna sono la vasocongestione pelvica, la lubrificazione e la dilatazione della vagina, e la tumescenza dei genitali esterni.
- Orgasmo. Questa fase consiste in un picco di piacere sessuale, con allentamento della tensione sessuale e contrazioni ritmiche dei muscoli perineali e degli organi riproduttivi. Nel maschio vi č la sensazione di inevitabilitŕ dell'eiaculazione, seguita dall'emissione di sperma. Nella femmina vi sono contrazioni (non sempre percepite soggettivamente come tali) della parete del terzo esterno della vagina.
- Risoluzione. Questa fase consiste in una sensazione di rilassamento muscolare e di benessere generale. Durante questa fase, i maschi sono fisiologicamente refrattari ad ulteriori erezioni ed orgasmi per un periodo variabile di tempo. Al contrario, le femmine possono essere in grado di rispondere a nuove stimolazioni quasi immediatamente.
I disturbi della risposta sessuale possono verificarsi in una o piů di queste fasi.
Sia nell'uomo che nella donna i disturbi della fase del desiderio sono il desiderio ipoattivo o
l'avversione sessuale.
Nell'uomo il disturbo piů comune della fase dell'eccitazione č il disturbo dell'erezione (impotenza),
mentre nella donna vi č la mancanza di eccitazione sessuale e di lubrificazione.
Nell'uomo il disturbo piů comune della fase dell'orgasmo č l'eiaculazione precoce, anche se esistono
uomini che hanno eiaculazione ritardata, impossibile o non piacevole, mentre nella donna č molto comune l'anorgasmia o frigiditŕ (impossibilitŕ di raggiungere l'orgasmo).
Esistono poi i cosiddetti disturbi da dolore sessuale, ovvero la dispareunia, sia maschile che femminile,
che consiste in un coito doloroso, solitamente dovuto a cause organiche, o il vaginismo femminile,
involontaria contrazione della vagina che impedisce la penetrazione.

|
 |
|