|
 |
|
|
|
|
|
|
|
Una malattia mitocondriale può colpire nell’età infantile come nell’età adulta ed è una malattia multisistemica. In quasi tutte le cellule del corpo ci sono delle piccole centrali dette mitocondri. La teoria vuole che 1,5 miliardi di anni fa, i mitocondri fossero cellule indipendenti, probabilmente dei batteri, e che siano stati inglobati in cellule di organismi superiori che necessitavano di una fonte rapida di energia. Quello che i mitocondri fanno per la cellula è una "rapida produzione di energia",ATP(ATP si può paragonare al denaro in contante). Qualsiasi cosa in grado di compromettere gravemente la produzione di ATP nei mitocondri può danneggiare o addirittura uccidere le cellule e causare così il malfunzionamento dei tessuti e l’apparizione dei sintomi. Il DNA mitocondriale viene ereditato esclusivamente dalla madre, attraverso i mitocondri del suo uovo; lo spermatozoo non dà nessun contributo. Successivamente ogni uovo e tutte le altre cellule del corpo sono dotate di centinaia di mitocondri, ed ogni mitocondrio può contenere molte molecole di DNA mitocondriale. Sebbene la cellula raddoppi approssimativamente il numero dei mitocondri e delle molecole mitocondriali prima di dividersi, la cellula originale non può regolare quali specifici mitocondri vadano ad ogni cellula figlia. Conseguentemente, se un uovo fertilizzato trasferisce una mutazione in qualche frazione del suo DNA mitocondriale (una condizione conosciuta come eteroplasmia) una cellula figlia può ereditare una proporzione più ampia di mitocondri mutati, e le altre cellule possono ereditare una percentuale più alta di mitocondri con DNA normale. Un figlio nato da un uovo eteroplasmico può quindi avere alcuni tessuti arricchiti di normale DNA mitocondriale o altri arricchiti di DNA mutante. Inoltre, gli ovuli di una donna con cellule eteroplasmiche possono differire nella percentuale di DNA mitocondriale mutante; i suoi figli possono quindi differenziarsi marcatamente nella estensione e distribuzione di molecole mutanti nei loro tessuti e nella loro gravità, e perfino nel tipo di sintomi che manifesteranno. Individui i quali si ammalano di una mutazione omoplasmatica, comunque, mostreranno tutti sintomi simili.
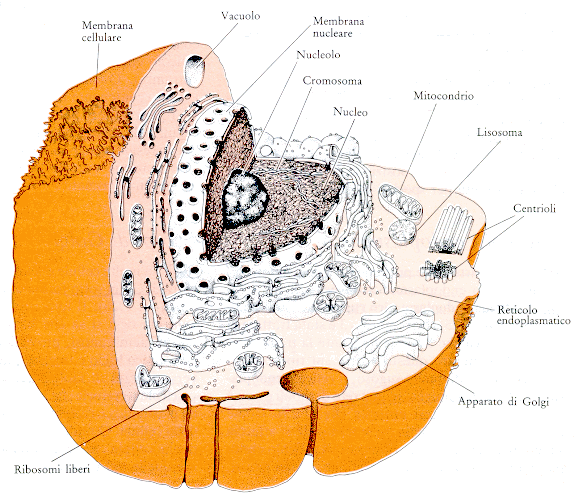
Membrana e Superficie cellulare Una sottile membrana, denominata membrana plasmatica, racchiude il contenuto di tutte le cellule viventi e costituisce una barriera fra l'ambiente intracellulare (interno) e quello extracellulare (esterno).
La membrana plasmatica è
costituita da un doppio strato continuo di molecole lipidiche, dello spessore di 8-10 nanometri (un nanometro
corrisponde a un miliardesimo di metro), attraversato parzialmente o completamente da numerose
proteine; essa funziona da barriera selettiva, regolando, così, la
composizione chimica della cellula.
Nucleo.
L'organulo più ingombrante della maggior parte delle cellule
vegetali e animali è il nucleo; si tratta di un corpuscolo delimitato da una
membrana, di forma e dimensioni variabili a seconda del tipo cellulare.
All'interno del nucleo sono conservati i
cromosomi, strutture filamentose composte da DNA e proteine e solitamente
presenti in coppie, in un numero variabile e caratteristico di ciascuna specie.
Il nucleo è delimitato da una doppia membrana, dotata di pori che
consentono le comunicazioni tra il nucleo e il resto della cellula (citoplasma).
Cromosomi: strutture filamentose composte da DNA e proteine e solitamente presenti in coppie, in un numero variabile e caratteristico di ciascuna specie. Mentre per gran parte del ciclo cellulare i filamenti cromosomali non sono distinguibili l'uno dall'altro, appena prima della divisione cellulare si ispessiscono e diventano visibili singolarmente. In ciascun cromosoma il DNA è presente sotto forma di una singola molecola, molto lunga e avvolta su se stessa a spirale, contenente una sequenza lineare di geni. I geni sono depositari dell'informazione necessaria per l'assemblaggio delle molecole indispensabili alla vita e alla riproduzione della cellula.
Citoplasta L'intero volume della cellula, con esclusione del nucleo, è occupato dal citoplasma. Esso contiene molte strutture e organuli. L'intero volume della cellula, con esclusione del nucleo, è occupato dal citoplasma. Esso contiene molte strutture e organuli specializzati che saranno descritti in seguito. La soluzione acquosa concentrata nella quale si trovano sospesi gli organuli cellulari è denominata citosol. Si tratta di un gel acquoso, contenente numerosissime molecole di varie dimensioni; nella maggior parte delle cellule esso è il compartimento cellulare di gran lunga più voluminoso (nei batteri è anche l'unico). Il citosol è il sito di molte funzioni importanti ai fini del mantenimento della cellula, compresi i primi stadi della demolizione delle molecole introdotte sotto forma di alimenti e la sintesi di numerose macromolecole che sono le unità costitutive della cellula. Sebbene molte molecole presenti nel citosol siano libere di muoversi per tutte le regioni della cellula, altre hanno una minore libertà di movimento poiché fanno parte di strutture ordinate che conferiscono al citosol un'organizzazione interna utile alle sue molteplici funzioni.
Membrana interna
Una rete tridimensionale di cisterne delimitate da membrane, denominata reticolo endoplasmatico, costituisce il compartimento cellulare dove avviene la sintesi di gran parte dei componenti delle membrane, come pure dei materiali destinati a essere esportati all'esterno della cellula.
Reticolo endoplasmatico rugoso
Il
reticolo endoplasmatico rugoso (RER) è una struttura cellulare costituita da un
sistema di pliche impilate e punteggiate di piccoli organelli detti ribosomi.
Apparato di Golgi Pile di cisterne appiattite, anch'esse delimitate da membrane, costituiscono, invece, l'apparato di Golgi, che riceve le molecole sintetizzate nel reticolo endoplasmatico, le elabora e le indirizza a diversi siti interni o esterni alla cellula. I lisosomi sono organuli piccoli, di forma irregolare, che contengono enzimi responsabili della digestione di numerose molecole inutili o nocive per la cellula. I perossisomi sono vescicole delimitate da membrana, che mettono a disposizione un ambiente isolato e circoscritto per reazioni nel corso delle quali vengono generate e demolite forme particolarmente pericolose e reattive dei perossidi di idrogeno. Inoltre, nella cellula vengono continuamente formate e distrutte piccole vescicole membranose, deputate al trasporto dei materiali da un organulo all'altro. In una tipica cellula animale il complesso degli organuli delimitati da membrana può occupare fino a metà del volume totale della cellula. sintesi proteica: Il nucleo controlla la sintesi proteica inviando nel citoplasma diverse molecole con funzione di messaggeri. I geni contenuti nel DNA vengono, infatti, copiati fedelmente all'interno del nucleo in un'altra molecola di acido nucleico, chiamata RNA messaggero (mRNA), che quando è matura passa nel citoplasma. Qui, interagendo con i ribosomi e con altre strutture molecolari, viene tradotto nella struttura primaria della specifica proteina per cui codifica il gene copiato originariamente.
Centrioli Un sistema di filamenti proteici, denominato citoscheletro, è presente nel citosol di tutte le cellule animali e vegetali. Nelle cellule animali, che mancano di una parete cellulare rigida, questo sistema ha un'importanza particolare, in quanto contribuisce a mantenere la struttura e la forma della cellula. Il citoscheletro fornisce un'impalcatura per l'organizzazione interna della cellula e un punto di ancoraggio per organuli ed enzimi. Esso permette, inoltre, alla cellula di compiere alcuni movimenti e, all'occasione, di cambiare forma. In molti tipi cellulari il citoscheletro è una struttura dinamica, che viene continuamente scomposta e riassemblata. È costituito da tre tipi principali di filamenti proteici: microtubuli, filamenti di actina e filamenti intermedi, connessi sia tra di loro che con altre strutture cellulari grazie a numerose proteine accessorie. Nelle cellule eucariotiche il movimento dipende per lo più dai filamenti di actina e miosina e dai microtubuli. Molte cellule possiedono sulla propria superficie strutture flessibili, simili a "peli", denominate ciglia o flagelli, contenenti un fascio centrale di microtubuli che funziona da motore del movimento. Ciglia e flagelli si piegano dando luogo a un battito regolare, simile a quello di una frusta, reso possibile dall'energia conservata all'interno dei microtubuli. I flagelli sono responsabili dei movimenti natatori degli spermatozoi; le ciglia che rivestono l'intestino o gli altri condotti presenti nell'organismo dei vertebrati indirizzano, con il loro movimento, i fluidi e le sostanze nutritive in una direzione particolare. Filamenti di actina, raccolti in grossi fasci, si trovano in tutte le cellule muscolari, dove, insieme a un'altra proteina, chiamata miosina, producono le tipiche contrazioni. Negli animali e nelle piante i movimenti associati alla divisione cellulare dipendono dai filamenti di actina e miosina e dai microtubuli, che guidano i cromosomi e gli altri componenti della cellula madre nelle due cellule figlie che si stanno formando.
Mitocondri e cloroplasti I mitocondri sono fra gli organuli più cospicui del citoplasma e sono presenti in quasi tutte le cellule eucariotiche. Essi hanno una struttura particolare, osservabile al microscopio elettronico: ciascun mitocondrio si presenta come un corpuscolo dalla caratteristica forma a fagiolo, lungo non più di 7 micrometri, delimitato da due membrane separate, la più interna delle quali presenta numerose pieghe (creste). (Lo studio, condotto da Rosario Rizzuto insieme a Paolo Pinton e Tullio Pozzan, in collaborazione con ricercatori americani dell’Università del Massachussetts, utilizzando un approccio tecnologico innovativo (ossia un microscopio a fluorescenza ultrarapido che permette di acquisire 30 immagini in meno di un secondo), ha ottenuto per la prima volta un’immagine tridimensionale di questi organelli in cellule viventi. I risultati di questo lavoro cambiano una nozione ormai consolidata in biologia: i mitocondri non sono infatti piccoli organelli “a forma di sigaro” distinti e indipendenti, come si era creduto finora, ma un intricato reticolo interconnesso, in rapido e continuo movimento all’interno della cellula. Questo nuovo concetto può servire a comprendere non solo processi importanti nella vita di questi organelli, ma anche come si sviluppano le malattie dovute a mutazioni del DNA mitocondriale. Inoltre è stato possibile dimostrare che i mitocondri sono a strettissimo contatto con un’altra struttura della cellula, il reticolo endoplasmatico, e grazie a questa vicinanza ricevono, al momento opportuno, il segnale di attivazione. Quando una cellula viene stimolata, infatti, il reticolo endoplasmatico rilascia ioni calcio nelle immediate vicinanze dei mitocondri, che li captano prontamente e vengono così attivati. Questo raffinato meccanismo di segnalazione e il ruolo fondamentale degli ioni calcio nel controllo della funzione mitocondriale aprono l’affascinante prospettiva di poterne “modulare” l’attività grazie allo sviluppo di nuovi farmaci che agiscano sul trasporto del calcio.) I mitocondri sono gli organuli responsabili della produzione di energia necessaria alla cellula per crescere e riprodursi; l'energia proviene dagli ultimi passaggi delle vie metaboliche che portano alla completa demolizione degli alimenti. Queste reazioni, che nel loro insieme costituiscono il processo di "respirazione cellulare", comportano il consumo di ossigeno e la produzione di anidride carbonica. In assenza di mitocondri molti organismi eucarioti non sarebbero in grado di utilizzare l'ossigeno per ricavare dagli alimenti tutta l'energia necessaria alle loro funzioni vitali. Diversamente dagli organismi aerobi, che non possono vivere in assenza di ossigeno, gli organismi anaerobi prosperano anche, o solo, in assenza di questo gas e le loro cellule sono prive di mitocondri.
I cloroplasti sono voluminosi organuli di colore verde, presenti solo nelle cellule delle piante e delle alghe, ma non negli animali o nei funghi. La loro struttura è ancora più complessa di quella dei mitocondri: oltre a essere avvolti da due membrane, all'interno presentano cisterne multiple, delimitate da una membrana contenente clorofilla (il pigmento verde delle piante). I cloroplasti svolgono un'importante funzione ecologica, in quanto sede della fotosintesi clorofilliana, che sfrutta l'energia dell'irradiazione solare per produrre ossigeno e molecole organiche a partire da semplici composti inorganici. L'ossigeno e le molecole organiche prodotte nei cloroplasti vengono poi utilizzate dalle cellule di altri organismi per la produzione di energia. Si calcola che tutto l'ossigeno presente nell'atmosfera sia derivato dall'attività fotosintetica dei cloroplasti.
I mitocondri sono formati da due membrane concentriche: una esterna, liscia, e una interna, ripiegata a formarIe varie creste. All'interno di questa membrana è contenuto un liquido denso (matrice) in cui sono immerse proteine di vario tipo. Le più importanti sono dette enzimi e servono a rendere più veloci, cioè a catalizzare, le diverse reazioni. Una volta ottenuto l'Atp, che è come una cassaforte piena di energia, per aprirla, è necessario l'intervento di un particolare enzima, detto Atpasi. Questo favorisce la rottura di legami chimici all'interno della molecola, liberando così l'energia che serve alla cellula. Ogni molecola di Atp fornisce ben 7,5 chilocalorie. I mitocondri hanno anche il privilegio (che condividono con i cloroplasti, presenti solo nelle cellule vegetali) di possedere una molecola di Dna autonoma da quella principale della cellula, presente nel nucleo. Inoltre, questo Dna mitocondriale ha forma circolare, come avviene solo nei batteri. " Ciò ha fatto pensare che i mitocondri derivino da un organismo autonomo, simile a una attuale cellula batterica, inglobato miliardi di anni fa da una primitiva cellula eucariote con la quale ha stabilito un rapporto di reciproco aiuto. La cellula infatti ottiene dai mitocondri l'energia per svolgere le sue funzioni, e nello stesso tempo gli fornisce i nutrienti necessari. Il Dna mitocondriale ha anche un'altra particolarità: si trasmette solo per via materna e per questo subisce un numero di mutazioni minori rispetto a quelle subite dal Dna principale.
Le proteine sono sostanze fondamentali per la vita degli organismi. Sono numerosissime e comprendono varie categorie. Nell'uomo, per esempio, sono proteine l'emoglobina, gli anticorpi, certi ormoni, i componenti dei muscoli, per ricordarne alcune. Ciascuna cellula di ogni organismo ha il compito di produrre determinate proteine, a seconda delle informazioni contenute nel suo Dna e dall'attivazione di alcuni geni piuttosto che di altri. Il processo di formazione è detto sintesi proteica e avviene nel reticolo endoplasmatico. Questo è un sistema di membrane, che contiene strutture tondeggianti dette ribosomi. Ed è proprio nei ribosomi che comincia la sintesi delle proteine. Si tratta di una specie di catena di montaggio. I ribosomi fabbricano un "semilavorato", che viene portato all'esterno attraverso le cosidette "vescicole di transizione", una specie di mezzo di trasporto che lo conduce in un altro sistema di membrane detto "apparato di Golgi" (dal nome del biologo Camillo Golgi, che per primo lo mise in evidenza nel 1898). " L'apparato di Golgi assomiglia a una pila di sacche appiattite a forma di disco, detta dittiosoma. Qui si ottiene il prodotto finito, cioè le proteine, che vengono successivamente "inscatolate" in vescicole di secrezione, che si distaccano dalla periferia delle sacche portando il loro contenuto fuori della cellula per distribuire le nuove proteine nell'organismo. " Oltre alla elaborazione di sostanze proteiche, l'apparato di Golgi svolge altre importanti funzioni. Innanzitutto, dalle sue vescicole hanno origine i lisosomi, cui è affidato il fondamentale compito di assicurare la "digestione cellulare". Questi elementi contengono infatti numerosi enzimi (idrolasi) specializzati nel degradare (ossia trasformare in molecole semplici) macromolecole e particelle provenienti dall'esterno della cellula. " Inoltre, come avviene per esempio nelle cellule vegetali, l'apparato di Golgi serve alla formazione di particolari sostanze: i polisaccaridi. Fra queste, l'amido, che è una riserva di energia contenuta nei semi, nelle radici e nei frutti, o la cellulosa, principale costituente della parete cellulare delle piante." Dividetevi e moltiplicatevi Mentre state leggendo questa frase, circa 100 milioni delle vostre cellule si sono divise, per moltiplicarsi. " Questo avviene con un processo, detto mitosi, nel corso del quale, partendo da un'unica cellula madre, si formano due cellule figlie geneticamente identiche, cioè con lo stesso numero di cromosomi. " Non tutte le cellule si moltiplicano o lo fanno sempre: dipende dai loro diversi compiti e dalle diverse necessità del tessuto o dell'organo che formano." Per esempio le cellule che costituiscono la nostra pelle, che ha bisogno di costanti ricambi, si dividono continuamente. Invece, i globuli rossi, elementi fondamentali del sangue, non si dividono mai. Il loro numero resta costante poiché quelli giunti alla fine della vita vengono sostituiti da altri nuovi prodotti dal midollo osseo." Una via di mezzo è quella delle cellule nervose, che dopo i primi anni di vita dell'individuo non si moltiplicano più. Anche le cellule del fegato abitualmente non si dividono, ma possono farlo all'occorrenza in caso di lesioni. " Il processo della mitosi inizia quando la membrana che avvolge il nucleo della cellulanucleare si dissolve, lasciando libero il Dna. " Nel frattempo la cellula si allunga, assumendo una forma affusolata, mentre al suo interno si costituisce una struttura con due poli, i centrioli, uniti da tante fibre. Nella regione centrale della cellula si vanno a disporre i cromosomi che costituiscono il Dna. Ognuno di essi si divide in due e ciascuna parte migra verso uno dei due poli." Nell'ultima fase del processo, si duplicano tutti gli altri elementi della cellula; nel frattempo la parte centrale comincia a restringersi fino a dividere la cellula in due. Si ricostituisce la membrana nucleare che va a racchiudere i cromosomi e si formano così le due cellule figlie." Ogni cellula deve sapere alla perfezione con quale ritmo dividersi e se dividersi oppure no: le cellule in cui salta il meccanismo che regola la loro proliferazione vanno incontro a una crescita incontrollata trasformandosi così in cellule tumorali. Il microchip della vita La cellula può essere considerata come un insieme di piccoli reparti di uno stabilimento chimico, ciascuno specializzato per una determinata funzione ma incapace di svolgerla senza la collaborazione degli altri. Tutto ciò è reso possibile da un continuo flusso di informazioni (che avviene sotto forma di uno scambio di molecole) col quale il suo funzionamento viene ottimizzato in accordo alle esigenze dell'organismo." Per rendere visivamente l'insieme di queste interazioni, si può pensare a uno schema simile a quello di un circuito integrato. Se consideriamo che ogni essere umano è composto da circa 200 tipi di cellule per un ammontare complessivo di centomila miliardi, e che tutte queste cellule sono in comunicazione tra loro e devono armonicamente interagire, si capisce la necessità di un continuo scambio di informazioni. " Essendo l'unità fondamentale della vita, ogni cellula deve essere capace di produrre le sostanze necessarie al proprio sviluppo e produrre nello stesso tempo energia; essere in grado di scambiare materia e energia con l'esterno; sapere come e quando farlo in base alle informazioni contenute nel Dna. Il meccanismo di regolazione all'interno della cellula dipende dalla precisione della consegna di queste informazioni, che tiene conto "in tempo reale" delle variazioni biochimiche avvenute nel micro ambiente circostante." Proprio come avviene in un chip, che deve elaborare, smistare e controllare le informazioni sulla base dei diversi comandi che gli vengono impartiti volta per volta dal programma. "
Informazioni generali
A volte, le malattie mitocondriali, sono così gravi da risultare incompatibili con la vita,
in altri casi danno solo disturbi lievi o impercettibili; nella maggior parte
della popolazione, invece, contribuiscono all'insieme dei fenomeni che
caratterizzano la senescenza. Le malattie mitocondriali, scoperte da poco più di
un decennio, sono diventate subito stelle di prima grandezza nel firmamento
delle patologie a eziologia ignota e sono entrate con prepotenza anche tra i
mali di cui si pensava di aver capito già molto, quali il cancro o il diabete. Troppo pochi dieci anni di studi
Infatti, anche se è noto fino dal 1963 che i mitocondri hanno
geni propri racchiusi in molecole circolari presenti in molte copie in ogni
mitocondrio, solo alla fine degli anni ottanta sono giunte le prime
dimostrazioni di un collegamento tra alcune neuro e miopatie ereditarie e
un'alterazione di questo particolare DNA. Queste scoperte sono principalmente
opera di Douglas Wallace, direttore del Centro di medicina molecolare della
Emory University di Atlanta, e di Salvatore Di Mauro, direttore del Dipartimento
di malattie neuromuscolari della Columbia University di New York. Fino a quel
momento, i malati hanno continuato a vagabondare da un reparto di neurologia
all'altro non solo senza che nessuno fosse in grado di curarli, ma anche senza
che fosse possibile fornire loro una spiegazione della propria condizione. Indizi difficili da decifrareA poco più di un decennio dalle prime scoperte, sono centinaia le malattie in cui è stato riscontrato un difetto del DNA mitocondriale, molte delle quali rarissime e altre assai comuni quali il diabete, il morbo di Parkinson e quello di Alzheimer e il cancro (vedi riquadro). Ma come riconoscerle? Nelle patologie più tipicamente mitocondriali, l'esordio molto spesso avviene attraverso un deficit visivo, che deriva da una degenerazione della retina - non a caso la retinite pigmentosa è presente in molte di esse - o del nervo ottico, oppure attraverso una manifestazione neurologica o neuromuscolare, o con entrambi gli eventi. «In letteratura sono descritti i casi di alcune famiglie con uno spettro di malattia davvero ampio» spiega Sciacco. «Con ogni probabilità esiste un valore soglia per cui in una stessa famiglia alcuni membri, che non lo raggiungono, hanno disturbi di poco conto, mentre i fratelli che passano il limite a volte non sopravvivono, oppure sono malati molto gravi. Ma non c'è una regola valida per tutti: ci è capitato di avere un paziente di ottant'anni che era sempre stato bene e che a un certo punto ha iniziato ad avere una ptosi palpebrale bioculare, che è rimasta l'unica manifestazione della sua malattia mitocondriale». Per orientarsi e per capire per tempo che quello che non va è nel piccolo anello di DNA delle minuscole dinamo cellulari è quindi indispensabile eseguire una serie di indagini strumentali e di laboratorio, oltre all'osservazione clinica. «Nel 40 per cento dei pazienti si riscontra un aumento della creatinfosfochinasi sierica, e in quasi tutti vi sono livelli di acido lattico molto alti, sia a riposo sia dopo uno sforzo» ricorda la neurologa del Policlinico. «Inoltre l'elettromiografia, l'elettroencefalogramma e la TC consentono di inquadrare la situazione e di tenerla sotto controllo. Ma ciò che forse è di maggior aiuto è l'esame istologico: nei tessuti interessati i mitocondri sono enormi oppure in numero molto superiore alla media. Se poi si fa una biopsia muscolare spiccano, in caso di positività, quelle che vengono chiamate le fibre stracciate o ragged red, forse la caratteristica più tipica delle malattie mitocondriali. Il tutto viene poi completato da un'analisi genetica». Pochi centri specializzati in Italia
Purtroppo, però, sono ancora poche le strutture dove vengono
eseguite tali biopsie, e il Centro del Policlinico è un riferimento per molti
medici del Nord Italia, che inviano lì i pazienti per una conferma diagnostica
(il riferimento per il Centro sud è il gruppo di Pietro Tonali del Policlinico
Gemelli, di Roma). «Al nostro centro giungono i malati provenienti dagli
ospedali con cui siamo collegati, e non solo. Per alcuni il riconoscimento è
stato abbastanza rapido, mentre molti altri hanno subito non poche peripezie
prima che qualcuno pensasse ai mitocondri» riferisce Monica Sciacco. Ed è
proprio grazie al fatto che a Milano arrivano molti pazienti che i neurologi
sono riusciti a studiare più di 200 di loro, identificando ogni singola
mutazione e cercando di collegarla al quadro clinico e biochimico delle diverse
malattie.
Incontro con Salvatore Di Mauro
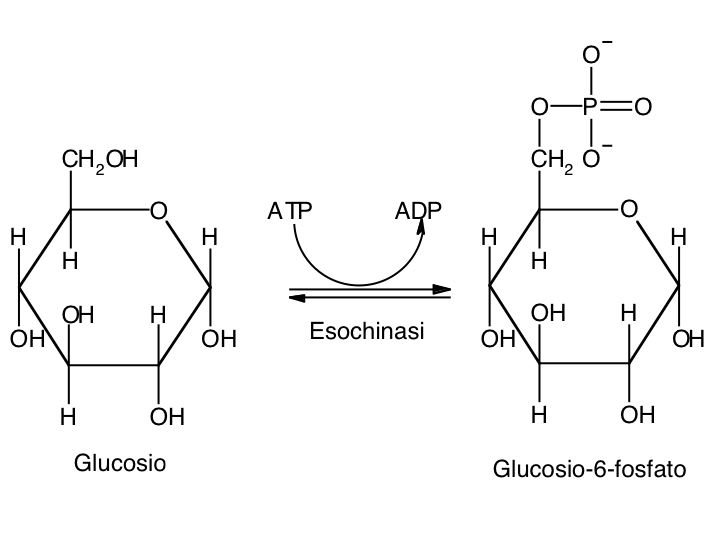
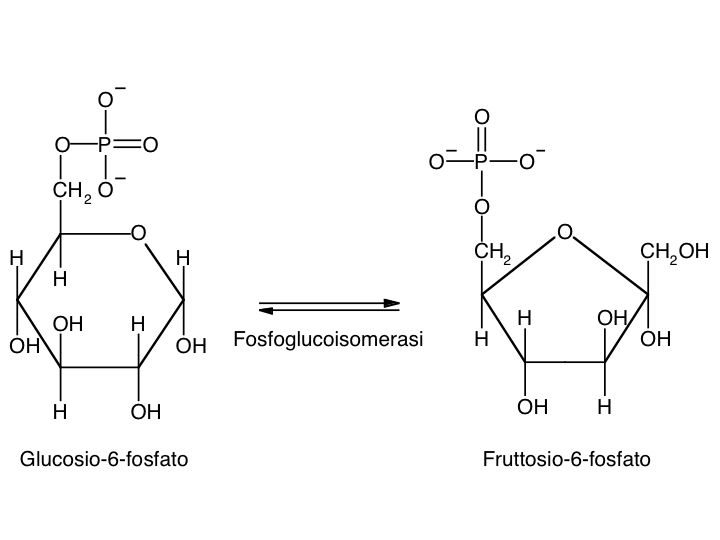
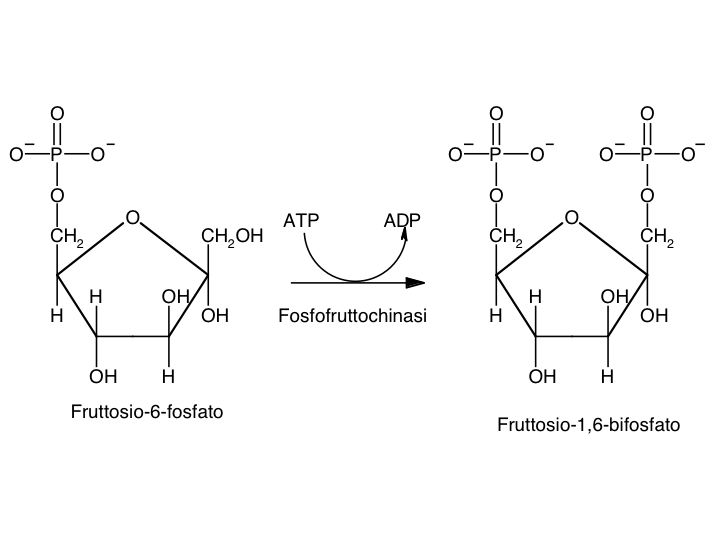
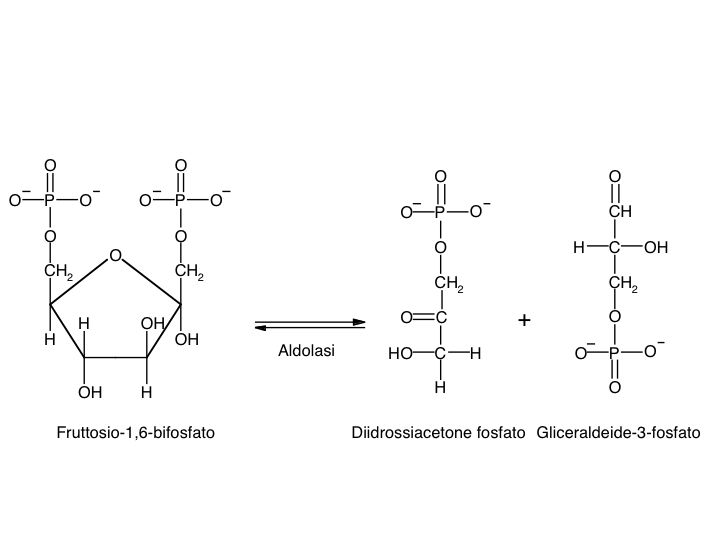
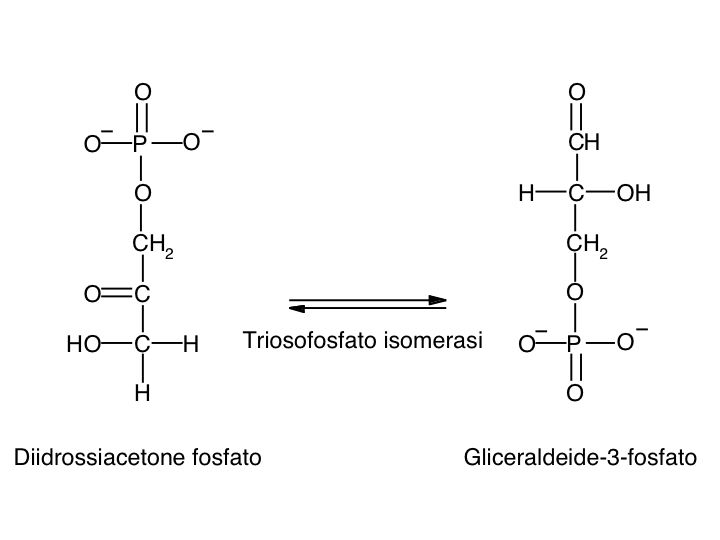
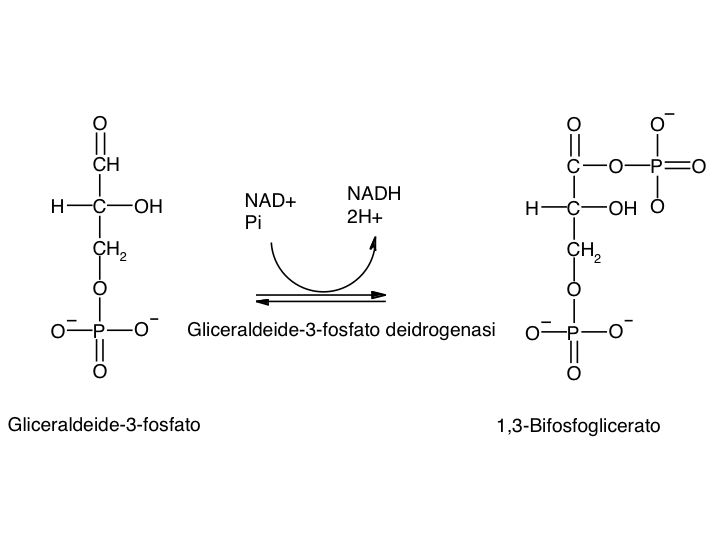
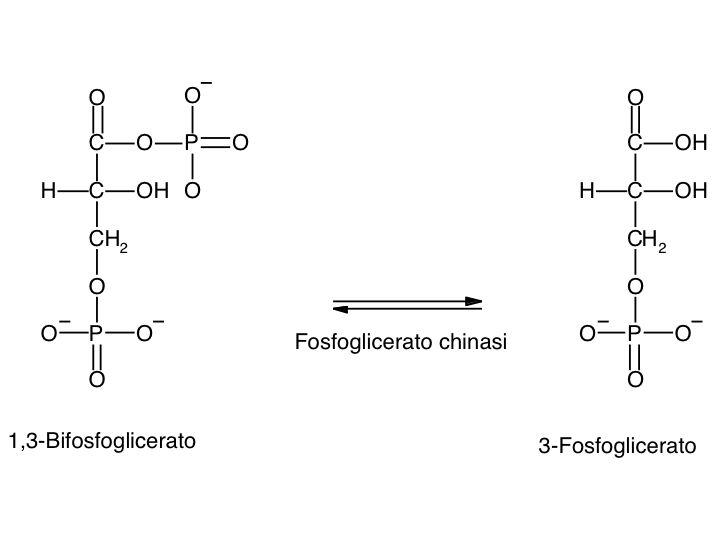
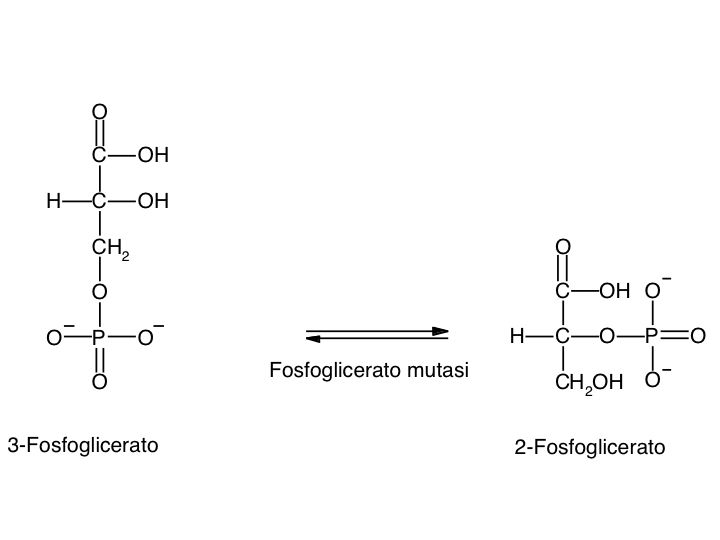
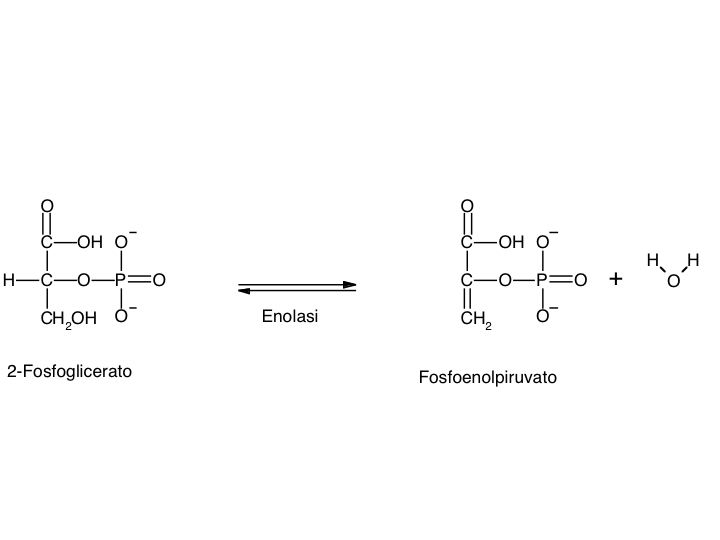
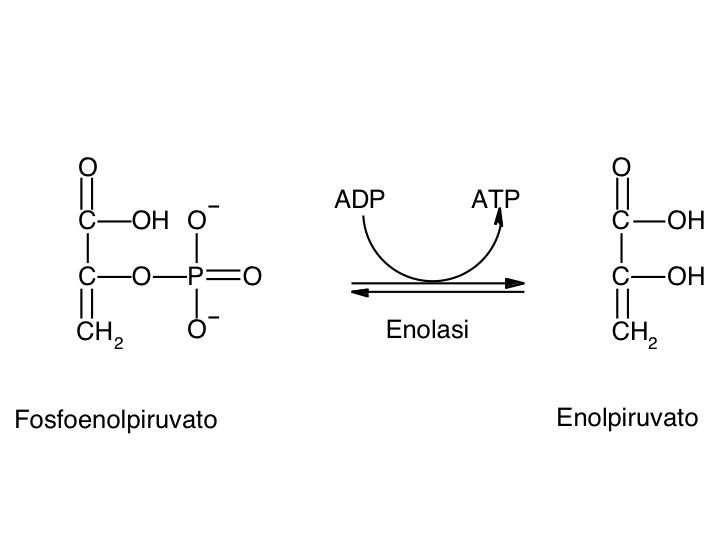
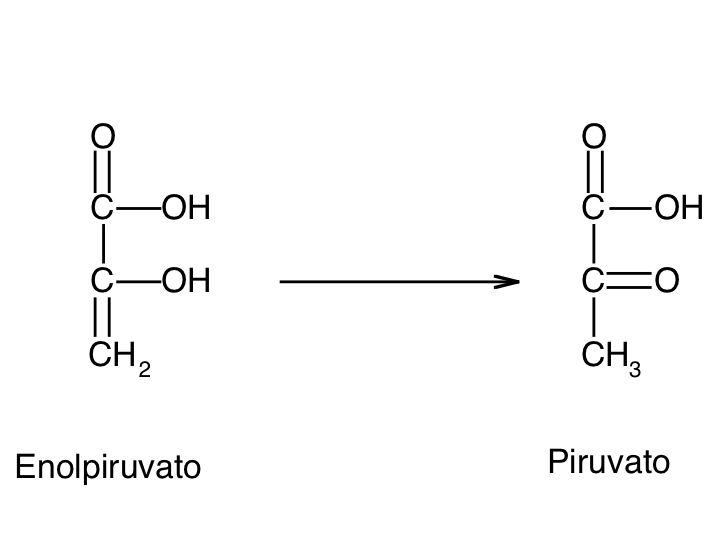
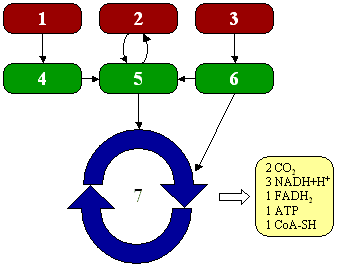
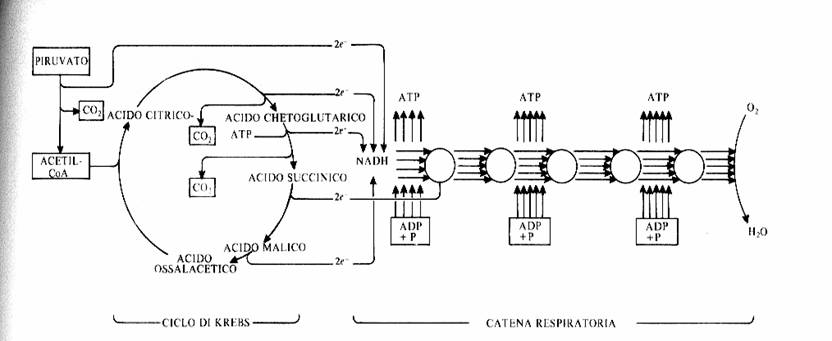
Le tre fai della respirazione cellulare La respirazione ha luogo in tre fai: glicolisi, ciclo di Krebs, catena respiratoria; la prima fase ha luogo nel citoplasma,le altre due nei mitocondri. Nelle prime due fasi gli atomi di idrogeno vengono staccati dal carbonio e trasferiti a composti detti accettori di elettroni. Gli accettori di elettroni che operano nella glicossi e nel ciclo di Krebs sono il NAD(nicotinide-adenin-dinucleotide) e il FAD (flavin-adenin-dinucleotide); ambedue hanno una struttura nucleotidica, come l’ATP. Sia il NAD che il FAD hanno la proprietà di accettare due atomo di ifrogeno. Nella terza fase della respirazione ha luogo un trasporto di elettroni da livelli energetici più alti a livelli energetici più bassi. Il processo si compie mediante passaggi sucessivi attraverso una serie di composti trasportatori di elettroni, detti citocromi. I citocromi hanno la struttura chimica simile a quella dell’emoglobina: un anello porfirinico al quale è unita una lumga coda proteica. La serie di citocromi costituisce la catene respiratoria, che è localizzata nei mitocondri ed è collegata agli enzimi che operano la ricarica dell’ADP, trasformandolo in ATP. Glicolisi Durante la glicolisi la molecola del glucosio, a 6 atomi di carbonio,, viene scissa in 2 atomi di piruvato, conposto da 3 atomi di carbonio: la scissione del glucosio ha luogo mediante una serie di 9 reazioni distinte, che costituiscono altrettante tappe durante le quali 2 molecole di ATP si trasformano in ADP e 4 atomo di idrogeno vengono trasferiti al NAD.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Glicolisi
|
2 NADH -------> 6 ATP ] ------------> 8 ATP 2 ATP |
|
Da piruvato ad acetil-CoA |
1 NADH -------> 3 ATP -------------> (x2) 6 ATP
|
|
Ciclo di Krebs
|
3 NADH -------> 9 ATP 1 FADH -------> 2 ATP ] ---------> (x2) 24 ATP
1 ATP |
ATP
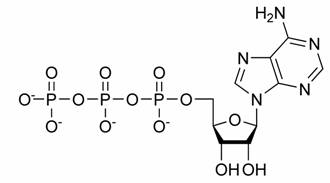
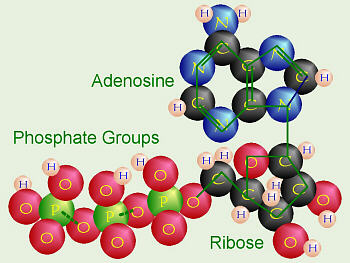
O=ossigeno (8)
P=Fosforo (15)
N=Azoto (7)
H=idrogeno (1)
C=carbonio (6)
Amminoacidi o aminoacidi
Le proteine alimentari, a
parte alcune eccezioni, non sono utili in quanto tali, ma come fonte di
amminoacidi. Infatti l'organismo scinde tramite la digestione le proteine
alimentari negli amminoacidi che le costituiscono, per poi ricostruire le
proprie proteine e altre molecole di importanza biologica.
Inoltre non tutti i venti amminoacidi sono necessari, ma solo nove, poiché
gli altri possono essere sintetizzati dall'organismo a partire da altre
sostanze.
Gli aminoacidi non servono solo nell'anabolismo muscolare; alcuni di essi vengono anche utilizzati a fini energetici. Il processo (gluconeogenesi) per il quale alcuni aminoacidi vengono trasformati in glucosio (poi usato come fonte energetica) dipende dall'intensità del lavoro e dalla durata: in un lavoro leggero di circa 40' solo il 4% dell'energia proviene dalle proteine, mentre in un lavoro intenso della stessa durata si arriva a un contributo del 15%; dopo 4 ore di lavoro leggero ben il 45% del glucosio liberato dal fegato proviene dalle proteine. Particolare importanza per il processo che ottiene energia dalle proteine rivestono i tre aminoacidi ramificati, (isoleucina, leucina, valina), così chiamati per la loro struttura.
Amminoacidi essenziali
Gli amminoacidi che
l'organismo non è un grado di autoprodurre si chiamano essenziali e sono
in tutto 8: triptofano, fenilalanina, lisina,
treonina, valina, leucina, isoleucina e valina.
I neonati non sono in grado di sintetizzare anche arginina e
istidina.
Il fabbisogno di amminoacidi essenziali è massimo durante i primi mesi di
vita, e diminuisci con l'età in quanto l'organismo diventa sempre più
efficiente nel riciclare gli amminoacidi essenziali. Anche il fabbisogno
di amminoacidi totali (e quindi di proteine alimentari) diminuisce con
l'età, ma in misura minore rispetto a quello di amminoacidi essenziali.
Gli amminoacidi ramificati sono: la leucina, l'isoleucina e la valina.
C'è chi sostiene che gli integratori di BCAA (ramificati) bypassino il metabolismo epatico per arrivare direttamente nei muscoli.
I ramificati hanno funzioni energetiche, aumentano la resistenza muscolare, hanno un'azione anticatabolica.Se presi dopo le sessioni di allenamento, velocizzano di conseguenza il recupero.
C'è chi sostiene che gli integratori di BCAA (ramificati) bypassino il metabolismo epatico per arrivare direttamente nei muscoli.
Consiglio l'utilizzo degli aminoacidi ramificati negli allenamenti dimagranti, negli allenamenti di definizione muscolare, negli allenamenti dove prevale la resistenza muscolare.
Dosaggi dei BCAA: 10-15 grammi a stomaco vuoto con 10 grammi di miele, metà prima dell'allenamento e l'altra metà subito dopo.
la fase anticatabolica di definizione in cui si segue una dieta ipocalorica per perdere tutto (o più) il grasso guadagnato nella precedente fase. Assieme a questa perdita di grasso vi è anche una perdita di massa magra, questa perdita dovrebbe essere minima e si dovrebbe limitare ad un rapporto di 2:1 (1 Kg di massa magra persa ogni 2 Kg di grasso perso).
L’anabolismo è il processo per cui una cellula viene “costruita”, partendo dai nutrienti semplici ottenuti dal suo ambiente. Poiché l’anabolismo ha come risultato la sintesi biochimica di nuovo materiale, è spesso chiamato biosintesi.
Il catabolismo è il processo per cui le sostanze chimiche vengono degradate e l’energia rilasciata
Il metabolismo è il risultato complessivo delle reazioni anaboliche e cataboliche.
Viene definita gluconeogenesi la produzione di glucosio partendo da precursori non saccaridici.
La gluconegenesi serve per sopperire alle mancanza di energia
La gluconeogenesi avviene
prevalentemente nel fegato e, in minima parte, nella corteccia renale.
La gluconeogenesi è energeticamente costosa, la reazione che consente di
sintetizzare glucosio partendo dal piruvato
Per quanto riguarda il fenomeno della gluconeogenesi, e cioè di quel meccanismo per cui l'organismo produce zuccheri smantellando le proteine corporee, va detto che esso non insorge immediatamente ma solo dopo un digiuno totale prolungato per diversi giorni.
Prima che si consumino le riserve di grasso passa del tempo. Inoltre la produzione di zuccheri ed energia attraverso la lisi delle proteine (e quindi dei muscoli in assenza di cibi proteici) è un meccanismo che l'organismo utilizza solo dopo che viene messo proprio alle strette.
l'organismo sintetizza il glucosio a partire dagli aminoacidi, dai grassi e dai corpi chetonici, tramite la gluconeogenesi (sia gli aminoacidi che i grassi presentano carbonio, idrogeno ed ossigeno, necessari alla formazione dei carboidrati).
Tavola periodica degli elementi
|
Gruppo |
1 |
2 |
3 |
|
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
|
Periodo |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
1 H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2 He |
|
2 |
3 Li |
4 Be |
|
|
|
|
|
|
|
|
|
|
|
5 B |
6 C |
7 N |
8 O |
9 F |
10 Ne |
|
3 |
11 Na |
12 Mg |
|
|
|
|
|
|
|
|
|
|
|
13 Al |
14 Si |
15 P |
16 S |
17 Cl |
18 Ar |
|
4 |
19 K |
20 Ca |
21 Sc |
|
22 Ti |
23 V |
24 Cr |
25 Mn |
26 Fe |
27 Co |
28 Ni l |
29 Cu |
30 Zn |
31 Ga |
32 Ge |
33 As |
34 Se |
35 Br |
36 Kr |
|
5 |
37 Rb |
38 Sr |
39 Y |
|
40 Zr |
41 Nb |
42 Mo |
43 |
44 Ru |
45 Rh |
46 Pd |
47 Ag |
48 Cd |
49 In |
50 Sn |
51 Sb |
52 Te |
53 I |
54 Xe |
|
6 |
55 Cs |
56 Ba |
57 La |
* |
72 Hf |
73 Ta |
74 W |
75 Re |
76 Os |
77 Ir |
78 Pt |
79 |
80 Hg |
81 Tl |
82 Pb |
83 Bi |
84 Po |
85 At |
86 Rn |
|
7 |
87 Fr |
88 Ra |
89 Ac |
* |
104 Rf |
105 Db |
106 Sg |
107 Bh |
108 Hs |
109 Mt |
110 Ds |
111 Rg |
112 Uub |
113 Uut |
114 Uuq |
115 Uup |
116 Uuh |
117 Uus |
118 Uuo |
|
* Lantanidi |
58 Ce |
59 |
60 |
61 |
62 |
63 |
64 |
65 |
66 |
67 |
68 |
69 |
70 |
71 |
|
** Attinidi |
90 |
91 |
92 |
93 |
94 |
95 |
96 |
97 |
98 |
99 |
100 |
101 |
102 |
103 |
|
Serie chimiche della tavola periodica |
||||
|
Metalli alcalini |
Metalli alcalino terrosi |
Lantanoidi |
Attinidi |
Metalli del blocco d |
|
Metalli del blocco p |
Metalloidi |
Nonmetalli |
Alogeni |
Gas nobili |
|
|
Ultimo aggiornamento: 21-06-06