SINDROME DI TURNER
La disgenesia gonadica, o anche detta sindrome di Turner, è caratterizzata da amenorrea primaria, infantilismo sessuale, bassa statura, anomalie congenite multiple e gonadi a striscia bilaterali in individui con fenotipo femminile, portatori di svariati difetti a livello del cromosoma X. Questa malattia deve essere distinta dalla:
| disgenesia gonadica mista, nella quale sono presenti un testicolo monolaterale e una gonade a striscia controlaterale. | |
| disgenesia gonadica pura, nella quale gonadi a striscia bilaterali si associano a un normale cariotipo 46,XX o 46,XY a statura regolare e ad amenorrea primaria. | |
| sindrome di Noonan, una malattia autosomica dominante che interessa entrambi i sessi, caratterizzata da pterigio del collo, bassa statura, cardiopatia congenita, cubito valgo e altri difetti congeniti, nonostante la presenza di un cariotipo e di gonadi normali. |
L’incidenza della malattia è di circa 1 ogni 2500 neonate femmine.
La diagnosi può essere formulata alla nascita, qualora siano già evidenti le anomalie
caratteristiche, o più frequentemente alla pubertà quando, oltre a queste anomalie, si
riscontra l'amenorrea e l'insufficiente sviluppo sessuale. La disgenesia gonadica spiega
circa un terzo delle amenorree primarie, costituendone la causa più frequente. I genitali
esterni sono indubbiamente femminili, ma rimangono immaturi e non vi è sviluppo mammario,
se non dopo terapia con estrogeni. I genitali interni sono costituiti da tube di Falloppio
e utero infantili e da gonadi a striscia bilaterali situate nei legamenti larghi. Le
cellule germinali primordiali compaiono transitoriamente durante l'embriogenesi e
scompaiono in seguito a un'atresia precoce. Dopo l'età in cui dovrebbe avvenire la
pubertà, nelle gonadi non sono più riconoscibili né follicoli né cellule uovo, ma
solamente tessuto fibroso indistinguibile da quello dello stroma ovarico normale. Le
alterazioni somatiche associate interessano principalmente lo scheletro e il connettivo.
Nell'infanzia gli aspetti che fanno sospettare la malattia sono il linfedema delle
estremità, lo pterigio del collo, la bassa attaccatura dei capelli, le pliche cutanee
sulla parte posteriore del collo, torace a scudo con capezzoli ampiamente distanziati e
basso peso corporeo alla nascita. Inoltre, la facies è caratterizzata da micrognazia,
epicanto, orecchie sporgenti a bassa inserzione o deformi, bocca a pesce, ptosi. In metà
dei pazienti è presente brevità dei quarto metacarpo, mentre il 10-20% è affetto da
coartazione aortica. Nell'adulto la statura media supera raramente i 150 cm. Altre
alterazioni associate comprendono malformazioni renali, nevi pigmentati, ipoplasia
ungueale, tendenza alla formazione di cheloidi, ipoacusia percettiva, ipertensione
essenziale, malattie autoimmuni.
Nel 20% dei soggetti si associa ipotiroidismo. 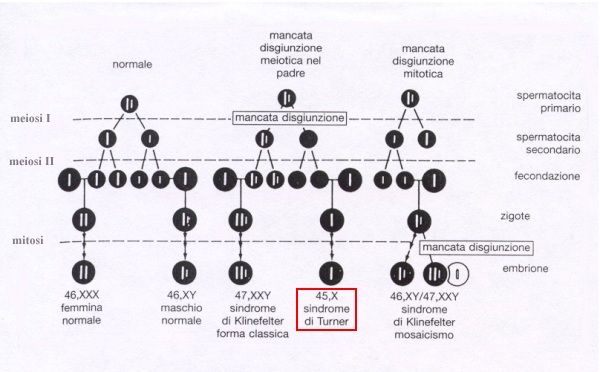
Circa la metà dei pazienti possiede un cariotipo 45,X, mentre circa un quarto di essi presenta un mosaicismo in assenza di anomalie strutturali (46,XX/45,X); la popolazione rimanente possiede un cromosoma X strutturalmente anomalo, con o senza mosaicismo. La variante 45,X può derivare sia dalla perdita, nel genitore, del cromosoma durante la gametogenesi, sia da un errore mitotico durante una delle suddivisioni precoci che avvengono nello zigote fecondato. La bassa statura e altri tratti somatici sono conseguenti alla perdita di materiale genetico dal braccio corto del cromosoma X. Una delezione a livello del braccio corto o del braccio lungo del cromosoma X dà luogo alla comparsa delle gonadi a striscia. Nei soggetti con mosaicismo o con alterazioni strutturali del cromosoma X, i fenotipi sono solitamente intermedi tra quello della variante 45,X e quello normale. In alcune pazienti con ipertrofìa del clitoride, oltre al cromosoma X, è stato rilevato il frammento di un cromosoma non identificato, probabilmente un cromosoma Y anomalo; nelle gonadi a striscia di questi pazienti si può sviluppare una neoplasia maligna. Talvolta la trasmissione familiare della disgenesia gonadica può essere conseguente alla traslocazione bilanciata tra il cromosoma X e un cromosoma autosomico. Lo studio del cariotipo è necessario per formulare la diagnosi e per identificare i soggetti con tracce di cromosoma Y e quelli con elevato rischio di tumore delle gonadi. Nel periodo in cui dovrebbe verificarsi la pubertà compaiono scarsi peli pubici e ascellari, le mammelle restano infantili e non compare il menarca. I livelli sierici di FSH sono elevati durante la prima infanzia, rientrano nella normalità durante la seconda infanzia e, intorno ai 9-10 anni, salgono ai livelli presenti nei soggetti castrati. Contemporaneamente aumenta anche l'LH sierico, mentre l'estradiolo plasmatico è basso [<40 pmol/l (10 pg/ml)]. Circa il 2% dei soggetti 45,X e il 12% di quelli con mosaicismo possiede un numero di follicoli residui sufficiente per permettere la comparsa delle mestruazioni. Sono stati anche riportati casi di gravidanza in donne con forme di malattia di modesta entità, tuttavia il periodo riproduttivo di queste donne è breve.
Si dovrebbe istituire una terapia sostitutiva con estrogeni nella fase prepuberale, allo scopo di indurre lo sviluppo delle manunelle, delle grandi e piccole labbra, della vagina, dell'utero e delle tube di Falloppio. Nel primo anno di trattamento con estradiolo l'accrescimento e la maturazione ossea raddoppiano; tuttavia i pazienti non raggiungono quasi mai un'altezza soddisfacente. La terapia combinata di oxandrolone e/o ormone della crescita accelera la crescita, ma non è stato stabilito se questa terapia abbia effetto sull'altezza finale. Nei soggetti 45,X sono rari i tumori gonadici, mentre sono frequenti nei soggetti con mosaicismo comprendente il cromosoma Y; per questo motivo si deve ricorrere all'asportazione degli abbozzi gonadici ogni volta che un paziente dimostra virilizzazione o presenta una linea cellulare comprendente il cromosoma Y.