Per
prima cosa impariamo a manipolare la molecola utilizzando il mouse.
Spostando il cursore nell'area nera, tenere il pulsante sinistro
premuto e muovere il mouse: noterete che la molecola ruota attorno ad
un ipotetico asse centrale. In questo modo sarà possibile
vedere
l'enzima sotto diversi punti di vista, e cercare la visione migliore.
Tenendo premuto il tasto SHIFT e muovendo il mouse col pulsante
sinistro premuto, si andrà a modificare lo zoom. Sempre
tenendo
premuto SHFT ma tenendo premuto il tasto destro, si ruoterà
la
molecola; infine, tenendo premuto CTRL e, contemporaneamente, anche il
tasto destro, riusciremo a spostare la molecola all'interno dell'area
nera.
Scaricando il file e visualizzandolo in una nuova finestra di Internet Explorer, nella barra di stato in basso appariranno le informazioni desiderate (nome dell'elemento, numero ordinale, amminoacido di appartenenza, numero ordinale di amminoacido, catena di appartenenza) riguardanti l'atomo che andremo a cliccare. Queste informazioni appaiono nella barra di stato in basso.
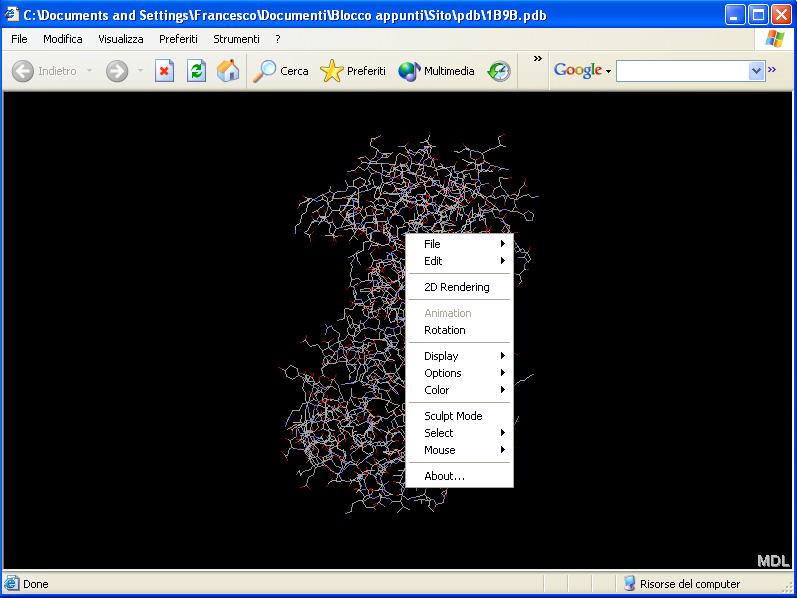
Ora che abbiamo capito i diversi modi in cui è possibile osservare una molecola, si può passare all'analisi delle diverse funzioni di Chime. Premendo col tasto destro, appparirà un menu a tendina con diverse opzioni:
Scaricando il file e visualizzandolo in una nuova finestra di Internet Explorer, nella barra di stato in basso appariranno le informazioni desiderate (nome dell'elemento, numero ordinale, amminoacido di appartenenza, numero ordinale di amminoacido, catena di appartenenza) riguardanti l'atomo che andremo a cliccare. Queste informazioni appaiono nella barra di stato in basso.
Ora che abbiamo capito i diversi modi in cui è possibile osservare una molecola, si può passare all'analisi delle diverse funzioni di Chime. Premendo col tasto destro, appparirà un menu a tendina con diverse opzioni:
Lasciamo
perdere le prime 4 e consideriamo direttamente la funzione "Rotation";
intuitivamente si capisce che cliccandoci sopra, la molecola
inizierà a girare lungo l'asse longitudinale alla
visualizzazione in cui la molecola si trova in quel momento. Durante la
rotazione è comunque possibile spostare, ingrandire, ruotare
l'immagine, esattamente agendo come visto sopra. Per bloccare la
rotazione sarà sufficiente deselezionare "Rotation".
Passiamo ora alla funzione successiva, ossia "Display". Chi conosce un minimo di biochimica strutturale saprà già che le proteine hanno diverse rappresentazioni. Spostando il mouse su "Display" si apre un sottomenu, in cui si potranno scegliere 8 visualizzazioni diverse:
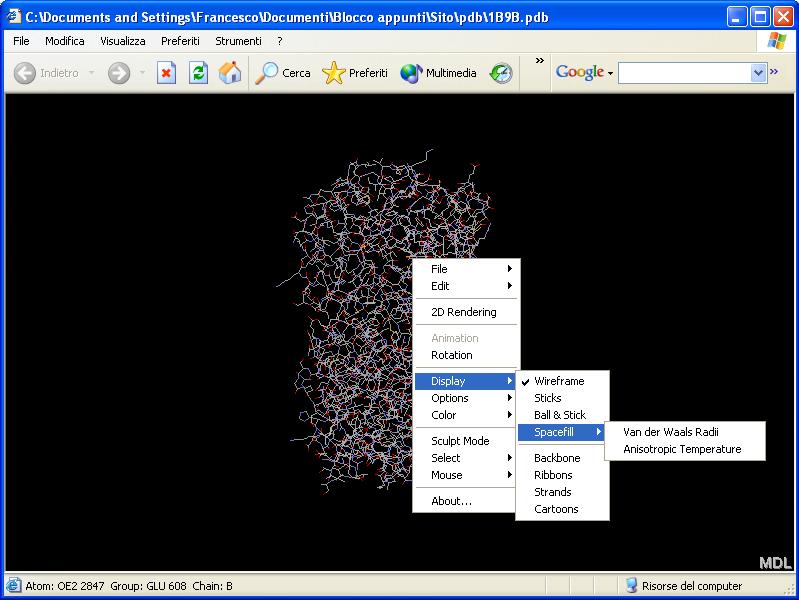
Passiamo ora alla funzione successiva, ossia "Display". Chi conosce un minimo di biochimica strutturale saprà già che le proteine hanno diverse rappresentazioni. Spostando il mouse su "Display" si apre un sottomenu, in cui si potranno scegliere 8 visualizzazioni diverse:
Bisogna
anche considerare che la visualizzazione "Spacefill" ha 2 ulteriori
possibilità di scelta, ma a noi ne interessa soltanto una.
Di
default, Chime visualizza una molecola in "Wireframe", ossia lo
scheletro della proteina, come potete osservare dal frame in alto. La
visualizzazione "Stick" permette di vedere lo scheletro della proteina
in versione "bastoncini" mentre il "Ball & stick" è
una
classica rappresentazione degli atomi come pallini, legati tra loro da
bastoncini. Tra le due visualizzazioni "Spacefill" disponibili, quella
che interessa maggiormente è "Van der Waals radii" che, come
suggerisce il nome, permette la visualizzazione degli atomi con i loro
rispettivi raggi di Van der Waals. Ai profani della materia, basti
sapere che questa visualizzazione permette di vedere gli atomi nella
loro dimensione reale.
Rimangono 4 visualizzazioni che mettono meglio in evidenza la struttura secondaria della proteina (per chiarimenti sulla struttura secondaria, consultate la pagina riguardante la struttura delle proteine). Tra le quattro, la più interessante è senza dubbio "Cartoons" che mostra molto charamente la disposizione di eliche e foglietti.
Vi consiglio, a questo punto, di provare tutte queste visualizzazioni sulla molecola di TPI in cima a questa pagina.
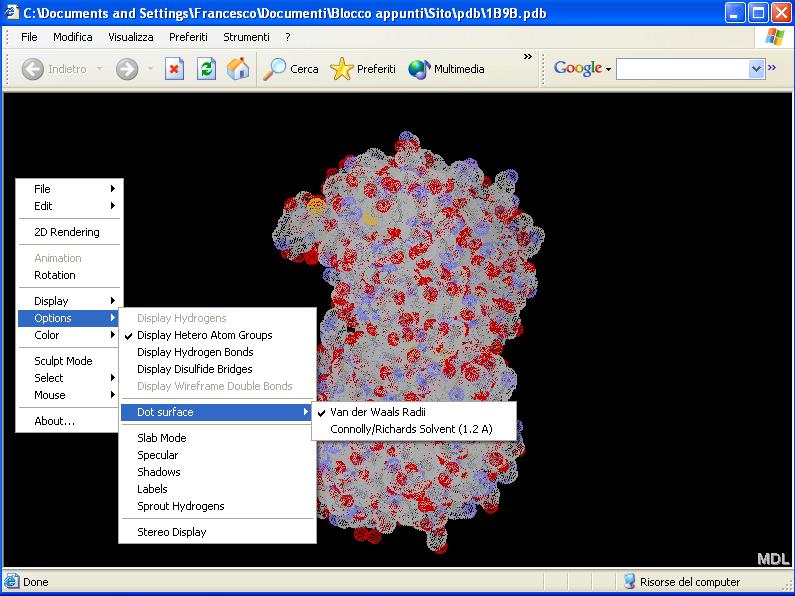
Rimaniamo nella visualizzazione "Sticks" e passiamo al menu successivo "Options". Ovviamente, vi consiglio vivamente di dare un'occhiata a tutte le funzioni ma quello che ci interessa è l'opzione "Dot surface" e la successiva scelta "Van der Waals radii", che permette una visualizzazione dei raggi di Van der Waals rappresentati come una nuvola di puntini attorno agli atomi:
Rimangono 4 visualizzazioni che mettono meglio in evidenza la struttura secondaria della proteina (per chiarimenti sulla struttura secondaria, consultate la pagina riguardante la struttura delle proteine). Tra le quattro, la più interessante è senza dubbio "Cartoons" che mostra molto charamente la disposizione di eliche e foglietti.
Vi consiglio, a questo punto, di provare tutte queste visualizzazioni sulla molecola di TPI in cima a questa pagina.
Rimaniamo nella visualizzazione "Sticks" e passiamo al menu successivo "Options". Ovviamente, vi consiglio vivamente di dare un'occhiata a tutte le funzioni ma quello che ci interessa è l'opzione "Dot surface" e la successiva scelta "Van der Waals radii", che permette una visualizzazione dei raggi di Van der Waals rappresentati come una nuvola di puntini attorno agli atomi:
Ora
andiamo su "Display" e passiamo alla visualizzazione "Van der Waals
radii". Ritorniamo al menu "Options" e scegliamo "Shadows". In questo
modo vengono mostrate le ombre, nei punti più nascosti;
questa
visualizzazione permette di individuare dei siti nascosti o poco
accessibili. "Labels" fa comparire delle etichette con informazioni
sopra ogni atomo.
Passiamo ora, dopo aver deselezionato "Shadows", ad analizzare il menu successivo, ossia "Color":
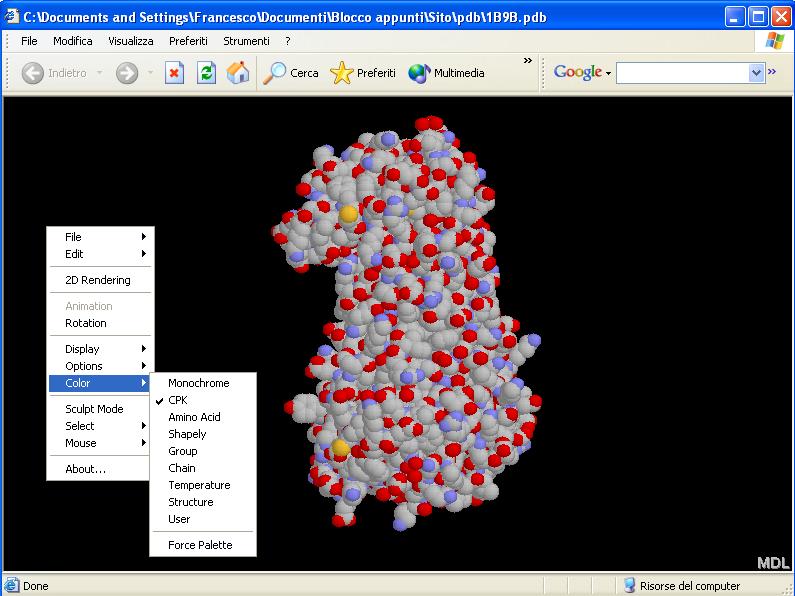
Passiamo ora, dopo aver deselezionato "Shadows", ad analizzare il menu successivo, ossia "Color":
Mi
sembra abbastanza semplice intuire la funzione di questo menu: da qui
sarà possibile cambiare i colori della molecola in
visualizzazione. Per visualizzare meglio questi cambiamenti, vi
consiglio di passare (se non lo siete già) alla
visualizzazione
"Van der Waals radii" anche se le modifiche apportate in questo menu
saranno comunque visibili anche nelle altre visualizzazioni; come
sempre, il mio consiglio è "PROVATE, PROVATE, PROVATE".
Di default, Chime visualizza la molecola colorata secondo "CPK" cambiando cioè colore in base agli atomi: i gruppi OH sono rossi (gli idrogeni non sono visualizzati), i carboni sono grigi, i gruppi NH sono celesti e i gruppi contenenti S sono gialli.
Ci si rende conto abbastanza facilmente che questa visualizzazione non è tra le più comode. "Monochrome", come suggerisce il nome, colora tutta la proteina di un solo colore, che è grigio di default ma che può essere modificato, come vedremo in seguito; "Amino acid" colora gli amminoacidi in maniera differente, in modo da poterli distinguere. Di "Shapely" non conosco la funzione esatta ma credo di aver capito che colora in maniera simile gli amminoacidi con caratteristiche simili (se qualcuno dovesse capire precisamente la funzione di quest'opzione, me lo facesse sapere. Gliene sarei molto grato). Delle opzioni "Group", "Structure" e "User" ignoro completamente la funzione. Per quanto riguarda "Temperature", non so in base a quale parametro vengano colorati i diversi residui amminoacidici ma mi sembra semplice capire che sia un parametro collegato alla temperatura. L'opzione credo più utilizzata sia "Chain", in grado di mettere in evidenza la struttra quaternaria della proteina. Anche qui rimando alla pagina sulla struttura delle proteine per chiarimenti. In questo modo vengono infatti colorate diversamente le varie catene polipeptidiche che formano la proteina nella sua forma quaternaria:
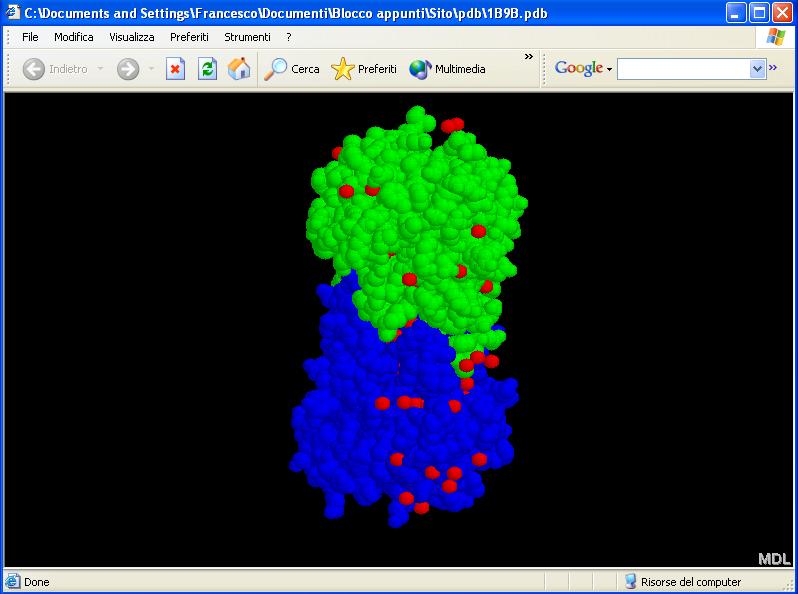
Di default, Chime visualizza la molecola colorata secondo "CPK" cambiando cioè colore in base agli atomi: i gruppi OH sono rossi (gli idrogeni non sono visualizzati), i carboni sono grigi, i gruppi NH sono celesti e i gruppi contenenti S sono gialli.
Ci si rende conto abbastanza facilmente che questa visualizzazione non è tra le più comode. "Monochrome", come suggerisce il nome, colora tutta la proteina di un solo colore, che è grigio di default ma che può essere modificato, come vedremo in seguito; "Amino acid" colora gli amminoacidi in maniera differente, in modo da poterli distinguere. Di "Shapely" non conosco la funzione esatta ma credo di aver capito che colora in maniera simile gli amminoacidi con caratteristiche simili (se qualcuno dovesse capire precisamente la funzione di quest'opzione, me lo facesse sapere. Gliene sarei molto grato). Delle opzioni "Group", "Structure" e "User" ignoro completamente la funzione. Per quanto riguarda "Temperature", non so in base a quale parametro vengano colorati i diversi residui amminoacidici ma mi sembra semplice capire che sia un parametro collegato alla temperatura. L'opzione credo più utilizzata sia "Chain", in grado di mettere in evidenza la struttra quaternaria della proteina. Anche qui rimando alla pagina sulla struttura delle proteine per chiarimenti. In questo modo vengono infatti colorate diversamente le varie catene polipeptidiche che formano la proteina nella sua forma quaternaria:
Se
vi state chiedendo cosa siano quei pallini rossi, beh vi dico che
è l'acqua. Le proteine, infatti, sono spesso circondate di
molecole d'acqua in quanto devono essere, in determinati punti,
altamente idratate.
A questo punto vi suggerisco di provare tutte queste colorazioni con tutte le diverse visualizzazioni, in modo da prendere dimestichezza con queste funzioni fondamentali.
Rimaniamo nella colorazione "Chain" e nella visualizzazione "Van der Waals radii" e, lasciando perdere l'opzione "Sculpt mode", passiamo all'opzione "Select", ossia quella che rende questo plug-in uno dei migliori visualizzatori gratuiti di molecole in 3D attualmente in circolazione:

A questo punto vi suggerisco di provare tutte queste colorazioni con tutte le diverse visualizzazioni, in modo da prendere dimestichezza con queste funzioni fondamentali.
Rimaniamo nella colorazione "Chain" e nella visualizzazione "Van der Waals radii" e, lasciando perdere l'opzione "Sculpt mode", passiamo all'opzione "Select", ossia quella che rende questo plug-in uno dei migliori visualizzatori gratuiti di molecole in 3D attualmente in circolazione:
Quello
che questo menu vi permetti di fare è di selezionare diverse
parti della molecola, in maniera selettiva, e di agire sulla parte
selezionata con i comandi visti sopra ("Display", "Options", "Color")
in maniera assolutamente indipendente dalle altre parti. Normalmente,
le modifiche fatte finora influivano su tutta la molecola, in quanto
Chime, di default, ha selezionata l'opzione "Select All" ma noi
possiamo fare in modo che certe modifiche agiscano solo su una parte
della molecola e altre su altre parti. Vediamo come.
Innanzitutto, l'opzione "Mouse click action" permette di stabilire l'azione che Chime deve svolgere quando clicchiamo su un atomo: "Identify" per identificarlo, "Distance" per calcolare la distanza tra 2 atomi cliccati, "Angle" per calcolare l'angolo formato da 3 atomi selezionati, "Torsion" per calcolare angoli di torsione (tutte queste informazioni appaiono nella barra di stato, come ho descritto prima), "Toggle atom label" per far apparire un'etichetta sull'atomo cliccato, in cui sono riportate informazioni che lo riguardano e, infine, "Pick center of rotation" che permette di far ruotare la molecola attorno all'atomo selezionato. Dell'opzione "Toggle distance monitor", sinceramente, ignoro la funzione:
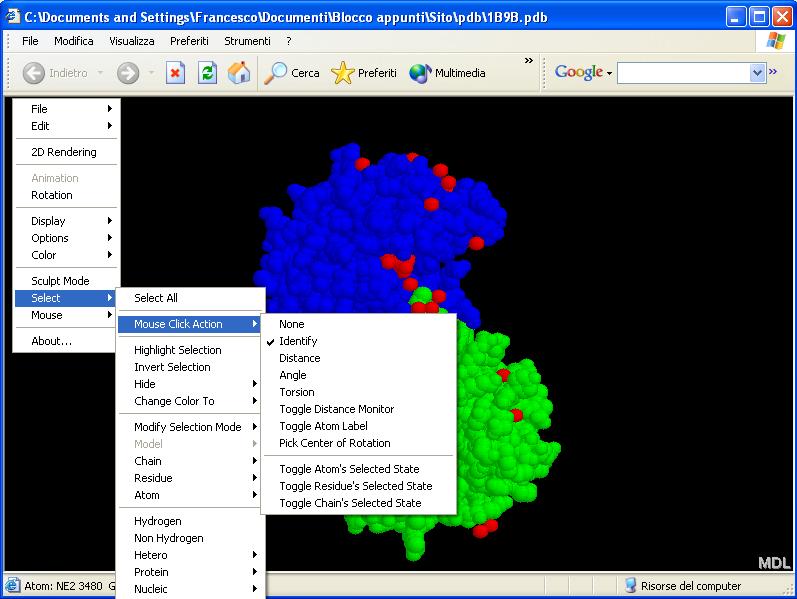
Innanzitutto, l'opzione "Mouse click action" permette di stabilire l'azione che Chime deve svolgere quando clicchiamo su un atomo: "Identify" per identificarlo, "Distance" per calcolare la distanza tra 2 atomi cliccati, "Angle" per calcolare l'angolo formato da 3 atomi selezionati, "Torsion" per calcolare angoli di torsione (tutte queste informazioni appaiono nella barra di stato, come ho descritto prima), "Toggle atom label" per far apparire un'etichetta sull'atomo cliccato, in cui sono riportate informazioni che lo riguardano e, infine, "Pick center of rotation" che permette di far ruotare la molecola attorno all'atomo selezionato. Dell'opzione "Toggle distance monitor", sinceramente, ignoro la funzione:
Per
il momento lasciamo selezionata l'opzione "Identify" e passiamo a
"Modify Selection
Mode", in cui decidiamo come Chime deve comportarsi di fronte ad una
nuova selezione: "Replace Selection" sostituisce la nuova selezione
alla precedente, "Add common to selection" aggiunge la nuova selezione
alla precedente, "Subtract from slection" toglie la nuova selezione dal
gruppo di selezione in cui era raggruppata, "Mutual exclusion" si
comporta come una normale funzione XOR, mutualizzando la selezione. Si
tratta comunque di una sezione che, personalmente adotto molte poche
volte.
Passiamo ora alle parti più importanti. "Chain" permette di selezionare una sola (o più, utilizzando le diverse modalità di selezione appena viste) catene polipeptidiche invece che l'intera proteina:
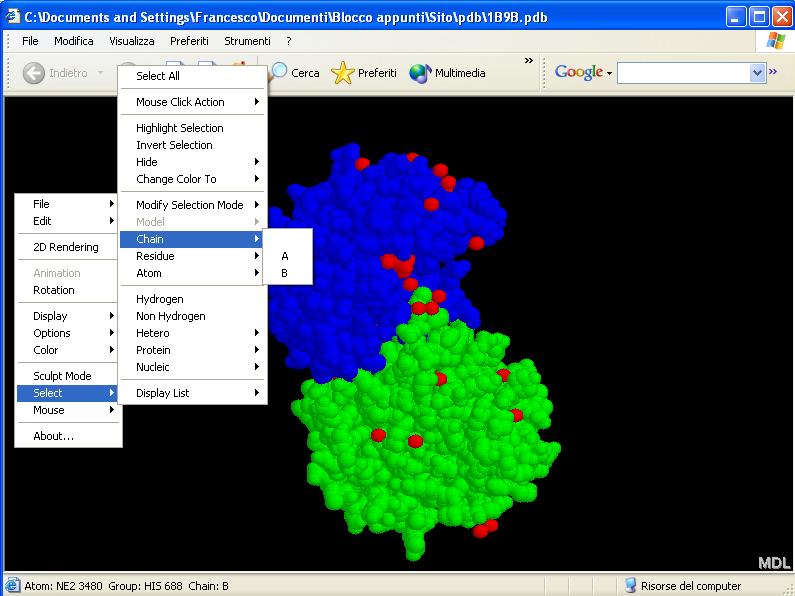
Passiamo ora alle parti più importanti. "Chain" permette di selezionare una sola (o più, utilizzando le diverse modalità di selezione appena viste) catene polipeptidiche invece che l'intera proteina:
Nel
nostro caso, la TPI ha solo 2 catene e quindi sarà possibile
usare la catena A o B separatamente. Selezionando la catena A, potremo
agire su di essa in maniera del tutto indipendente che da B: proviamo
ad andare su "Display" e selezioniamo "Cartoon". Notiamo come solo la
catena A venga visualizzata in foglietti ed eliche, mentre la catena B
rimane in visualizzazione "Van der Waals radii":
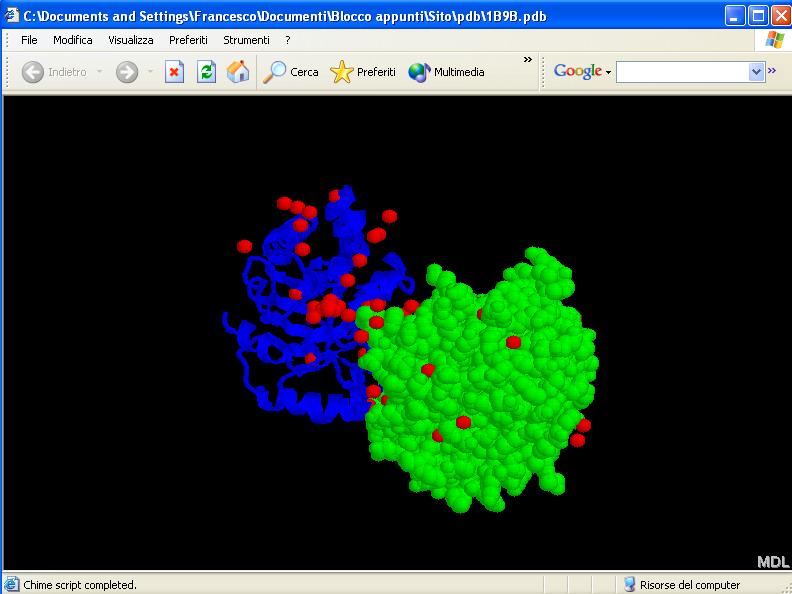
Possiamo
cambiare colore, opzioni e visualizzazione sepratamente per le diverse
catene.
Il menu successivo "Residue" permette di selezionare tutti gli amminoacidi di un solo tipo: in questo modo potremmo, ad esempio, mettere in risalto le leucine della catena A. Per fare questo, andiamo in Select e portiamoci su "Modify Selection Mode". Selezioniamo "Add common to Selection" e andiamo prima a selezionare la catena A ("Chain" - "A"), quindi su "Residue", selezioniamo "Leu". Ora portiamoci su "Display" e scegliamo "Stick":
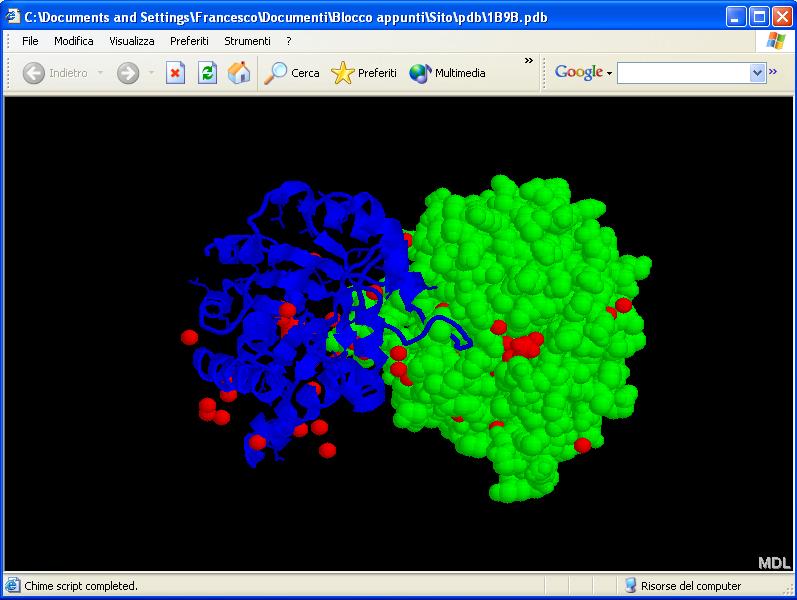

Il menu successivo "Residue" permette di selezionare tutti gli amminoacidi di un solo tipo: in questo modo potremmo, ad esempio, mettere in risalto le leucine della catena A. Per fare questo, andiamo in Select e portiamoci su "Modify Selection Mode". Selezioniamo "Add common to Selection" e andiamo prima a selezionare la catena A ("Chain" - "A"), quindi su "Residue", selezioniamo "Leu". Ora portiamoci su "Display" e scegliamo "Stick":
Potete
notare come la catena B sia rimasta immutata, mentre nella colonna A
siano state messe in evidenza le leucine.
Con la funzione "Atom" si possono selezionare direttamente tutti gli atomi di un tipo, ad esempio, tutti i C, tutti gli O, ecc.
Se, ad esempio, nella selezione corrente, noi andassimo a selezionare C e visualizzassimo "Van der Waals radii" in "Display", allora potremmo individuare tutti gli atomi di carbonio delle sole leucine della sola catena A. Se invece volessimo mettere in evidenza anche gli altri "C", allora dovremmo togliere l'opzione "Add to common selection" sostituendola con "Replace selction" (in questo momento abbiamo selezionato la catena A e le leucine, visualizzate in maniera differente). Andiamo a selezionare la catena A, perdendo così la selezione delle leucine. Ora ritorniamo a "Modify Selection" e riselezioniamo "Add to common selection" (se non lo facessimo, la successiva selezione degli atomi riguarderebbe anche la catena B), e selezioniamo separatamente "Chain" A e "Atom" C (in realtà, per poterlo visualizzare meglio, consiglio di selezionare un atomo meno frequente, come lo zolfo, S) e, su "Display" selezioniamo "Van der Waals radii". A questo punto avremo la catena B in "Van der Waals radii", la catena A in "Cartoons", le leucine della catena A in "Sticks" e tutti gli atomi di zolfo o carbonio della catena A in "Van der Waals radii":
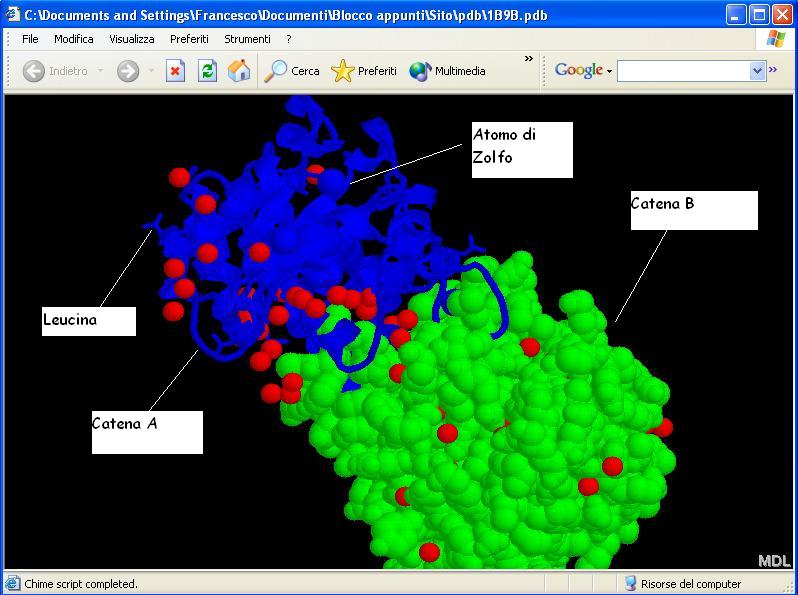
Con la funzione "Atom" si possono selezionare direttamente tutti gli atomi di un tipo, ad esempio, tutti i C, tutti gli O, ecc.
Se, ad esempio, nella selezione corrente, noi andassimo a selezionare C e visualizzassimo "Van der Waals radii" in "Display", allora potremmo individuare tutti gli atomi di carbonio delle sole leucine della sola catena A. Se invece volessimo mettere in evidenza anche gli altri "C", allora dovremmo togliere l'opzione "Add to common selection" sostituendola con "Replace selction" (in questo momento abbiamo selezionato la catena A e le leucine, visualizzate in maniera differente). Andiamo a selezionare la catena A, perdendo così la selezione delle leucine. Ora ritorniamo a "Modify Selection" e riselezioniamo "Add to common selection" (se non lo facessimo, la successiva selezione degli atomi riguarderebbe anche la catena B), e selezioniamo separatamente "Chain" A e "Atom" C (in realtà, per poterlo visualizzare meglio, consiglio di selezionare un atomo meno frequente, come lo zolfo, S) e, su "Display" selezioniamo "Van der Waals radii". A questo punto avremo la catena B in "Van der Waals radii", la catena A in "Cartoons", le leucine della catena A in "Sticks" e tutti gli atomi di zolfo o carbonio della catena A in "Van der Waals radii":
Vi
consiglio di passare molto tempo a girare queste opzioni
perchè
sono fondamentali: una volta presa pratica con queste tecniche di
logica abbastanza semplici, applicate al programma, vi renderete conto
delle reali potenzialitò di questo programma.
Con "Hetero" potrete selezionare tutto quello che non è strettamente collegato alla proteina, come solvente, acqua, ioni metallici usati come cofattori (vedi struttura delle proteine), ligandi, ecc.
Del menu "Protein" basta sapere che "Protein" seleziona tutta la parte proteica della visualizzazione (in caso siano presenti anche altre strutture non proteiche), "Alpha Carbon" seleziona solo i carboni alfa (vedi struttura delle proteine), "Backbone" individua tutti gli amminoacidi coinvolti in strutture secondarie, "Helix" quelli che formano alfa-eliche, "Sheet" quelli che formano foglietti-beta, "Sidechain" seleziona solo le catene laterali degli amminoacidi (vedi struttura delle proteine), "Surface" seleziona solo gli amminoacidi di superficie, "Acidic" gli amminoacidi acidi, "Acyclic" gli amminoacidi senza catene laterali cicliche, "Aliphatic" gli amminoacidi con catena laterale alifatica, "Aromatic" gli amminoacidi con catena laterale aromatica, "Charged" gli amminoacidi carichi, "Cyclic" gli amminoacidi ciclici (come la prolina) o con catena laterale ciclica, "Hydrophobic" gli amminoacidi idrofobici, "Neutral" gli amminoacidi neutri, "Polar" gli amminoacidi polari:
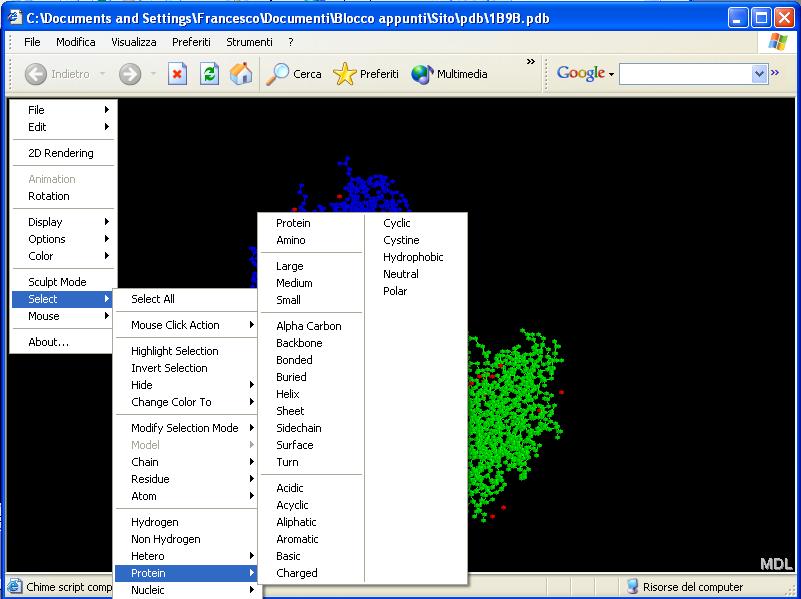
Con "Hetero" potrete selezionare tutto quello che non è strettamente collegato alla proteina, come solvente, acqua, ioni metallici usati come cofattori (vedi struttura delle proteine), ligandi, ecc.
Del menu "Protein" basta sapere che "Protein" seleziona tutta la parte proteica della visualizzazione (in caso siano presenti anche altre strutture non proteiche), "Alpha Carbon" seleziona solo i carboni alfa (vedi struttura delle proteine), "Backbone" individua tutti gli amminoacidi coinvolti in strutture secondarie, "Helix" quelli che formano alfa-eliche, "Sheet" quelli che formano foglietti-beta, "Sidechain" seleziona solo le catene laterali degli amminoacidi (vedi struttura delle proteine), "Surface" seleziona solo gli amminoacidi di superficie, "Acidic" gli amminoacidi acidi, "Acyclic" gli amminoacidi senza catene laterali cicliche, "Aliphatic" gli amminoacidi con catena laterale alifatica, "Aromatic" gli amminoacidi con catena laterale aromatica, "Charged" gli amminoacidi carichi, "Cyclic" gli amminoacidi ciclici (come la prolina) o con catena laterale ciclica, "Hydrophobic" gli amminoacidi idrofobici, "Neutral" gli amminoacidi neutri, "Polar" gli amminoacidi polari:
Combinando
queste diverse opzioni tra loro come abbiamo appena visto, si riesce a
mettere in evidenza l'aspetto desiderato.
"Nucleic" riguarda la messa in risalto di determinati aspetti di acidi nucleici, come DNA ed RNA: "Nucleic" mette in risalto tutta la parte nucleica della visualizzazione (nel caso di visualizzazione con strutture non nucleiche), "DNA" seleziona tutti gli elementi riguardanti il DNA, "Backbone" mette in evidenza lo scheletro dell'acido nucleico, "AT" evidenzia tutte le basi A e T appaiate, "CG" è la funzione analoga ad AT per le basi G e C, "Purine" evidenzia solo le basi di natura purinica, "Pyrimidine" evidenzia solo le basi pirimidiniche.
L'opzione "Change color to", sotto il menu "Select" permette di cambiare il colore della selezione corrente.
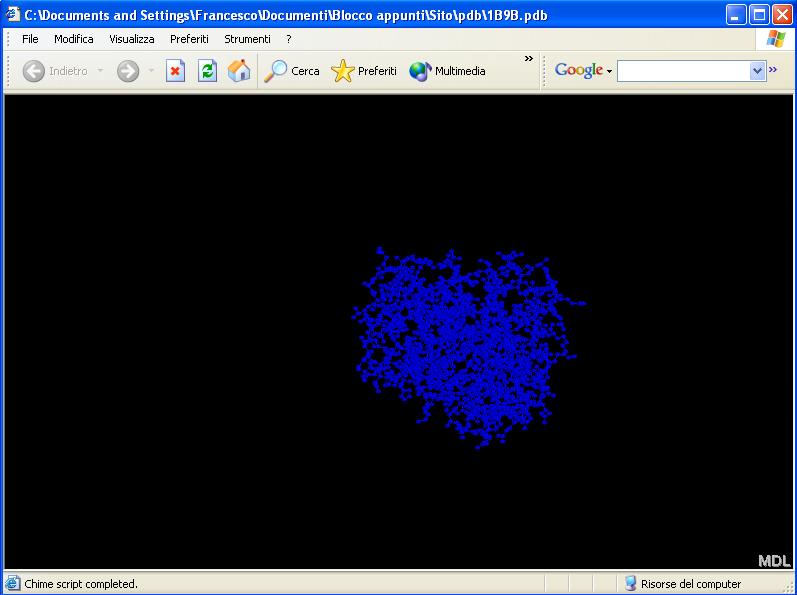
Un'ultima considerazione va fatta per le opzioni "Invert selection" e "Hide". Se usiamo "Invert selection" allora deselezioniamo l'elemento appena selzionato per selezionare tutto il resto: selezioniamo la catena A e scegliamo "Invert selection" per selezionare la catena B.
"Hide" apre un menu a più scelte: "Hide selected" permette di nascondere ciò che abbiamo appena selezionato (può essere usata per nascondere le molecole di acqua, che vengono raggruppate nella categoria "Hetero - Water") mentre "Hide not selcted" nasconde tutto quello che non è attualmente selezionato. In questo modo, si può, ad esempio, evidenziare solo una delle due catene di TPI: selezioniamo la catena A e scegliamo "Hide not selcted" per nascondere la catena B:
"Nucleic" riguarda la messa in risalto di determinati aspetti di acidi nucleici, come DNA ed RNA: "Nucleic" mette in risalto tutta la parte nucleica della visualizzazione (nel caso di visualizzazione con strutture non nucleiche), "DNA" seleziona tutti gli elementi riguardanti il DNA, "Backbone" mette in evidenza lo scheletro dell'acido nucleico, "AT" evidenzia tutte le basi A e T appaiate, "CG" è la funzione analoga ad AT per le basi G e C, "Purine" evidenzia solo le basi di natura purinica, "Pyrimidine" evidenzia solo le basi pirimidiniche.
L'opzione "Change color to", sotto il menu "Select" permette di cambiare il colore della selezione corrente.
Un'ultima considerazione va fatta per le opzioni "Invert selection" e "Hide". Se usiamo "Invert selection" allora deselezioniamo l'elemento appena selzionato per selezionare tutto il resto: selezioniamo la catena A e scegliamo "Invert selection" per selezionare la catena B.
"Hide" apre un menu a più scelte: "Hide selected" permette di nascondere ciò che abbiamo appena selezionato (può essere usata per nascondere le molecole di acqua, che vengono raggruppate nella categoria "Hetero - Water") mentre "Hide not selcted" nasconde tutto quello che non è attualmente selezionato. In questo modo, si può, ad esempio, evidenziare solo una delle due catene di TPI: selezioniamo la catena A e scegliamo "Hide not selcted" per nascondere la catena B:
Spero
di esservi stato utile per capire questo programma. Ho tralasciato
alcune funzioni, molte delle quali perchè, lo ammetto, non
ne conosco
neanche la funzione, altre perchè sono veramente poco
utilizzate. Consiglio vivamente a tutti di "giocare" il più
possibile con queste molecole, per poter capire a fondo il programma,
esattamente come ho fatto io qualche tempo fa.