Torna a ARGOMENTI
DI CHIMICA ORGANICA
LE REAZIONI DI SOSTITUZIONE ELETTROFILA AROMATICA
Il
meccanismo generale della reazione di sostituzione elettrofila aromatica al
benezene prevede nel primo stadio l’addizione dell’elettrofilo all’anello
benzenico e, nel secondo stadio, l’eliminzaione di un protone. Il risultato
finale è la sostituzione di un atomo di idrogeno con un altro gruppo:
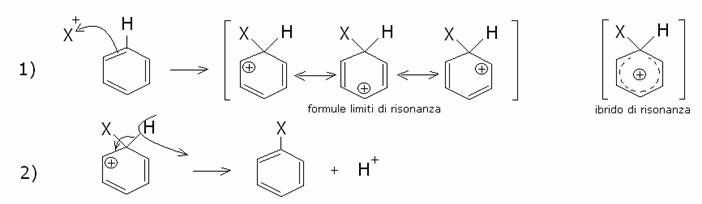
Le
principali reazioni di sostituzione elettrofila aromatica sono:
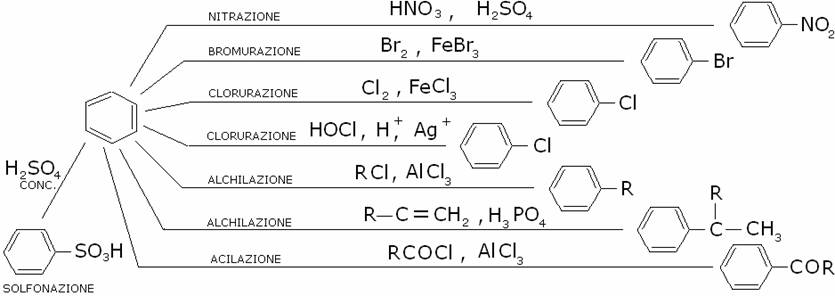
1) Nitrazione
Nel primo
stadio si forma l’elettrofilo:
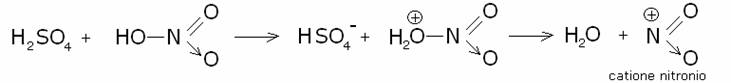
Il
catione nitronio attacca quindi l’anello
benzenico:
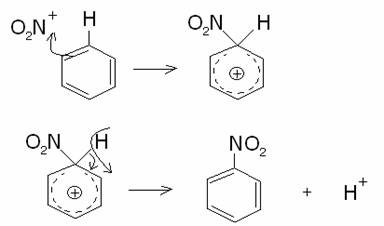
2)
Formazione di ammine aromatiche
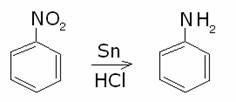
Il
nitrobenzene può essere ridotto ad amminobenzene (anilina) per riduzione con
stagno
metallico in
soluzione acida:
3) Alogenazione
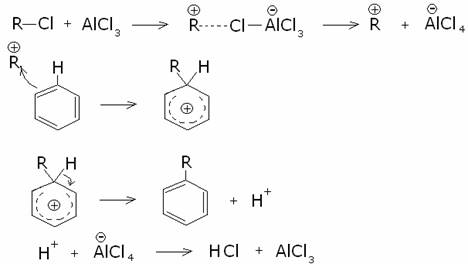
di Friedel-Kraft
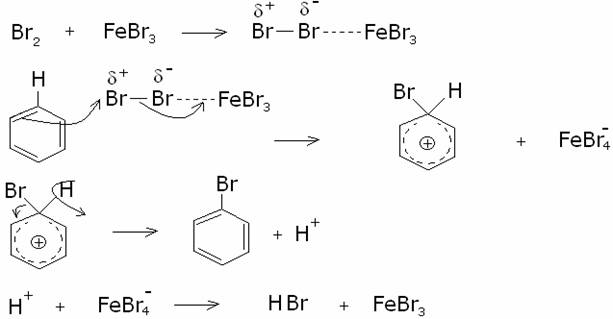
La reazione di
alogenazione richiede la presenza di un catalizzatore in grado di accettare
doppietti elettronici (acido di Lewis), generalmente alogenuri metallici: FeBr3
,AlCl3 , ZnCl2.
Meccanismo di reazione:
L’ordine
di reattività degli alogeni è: F2>Cl2>Br2>I2.
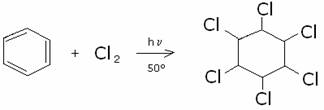
Durante la reazione di alogenazione, il sistema deve essere protetto dalla luce
solare, poiché possono verificarsi addizioni radicaliche all’anello benzenico:
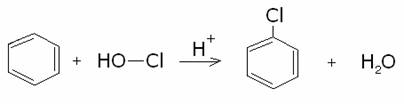
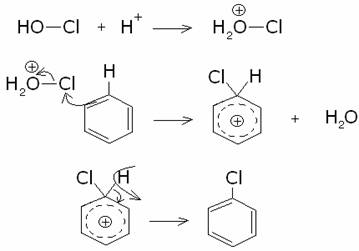
L’alogenazione
può avvenire anche tramite reazione del benzene con un acido ipoalogenoso, in
soluzione con un acido forte:
Meccanismo:
4)
Alchilazione
Il benzene
reagisce con un alogenuro alchilico, in presenza di tricolruro di alluminio
come catalizzatore, per dare un alchilbenzene. Il meccanismo è simile a quello
dell’alogenazione:
Questo metodo per ottenere un alchilbenzene può essere
utilizzato con vantaggio se il gruppo alchilico da legare al benzene è un
metile o un etile. Per radicali più complessi c’è il rischio di avere miscele
di alchilbenzeni a causa della possibilità del catione R+ di
riarrangiarsi in un catione più stabile. Ad esempio, utilizzando il cloruro di
propile, oltre al propilbenzene (o 1-fenil propano), si forma anche
l’isoprpilbenzene (o 2-fenil propano) per riarrangiamento del catione
propilico, primario, a catione isopropilico, secondario (più stabile), per
trasposizione di uno ione idruro:
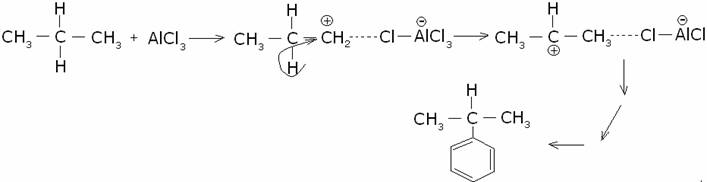
L’alchilazione
può effettuarsi anche con alcoli o alcheni in presenza di catalizzatori acidi
come acido fosforico, acido solforico, acido fluoridrico, trifluoruro di boro
(acido di Lewis):

Nel
primo stadio l’alchene viene protonato. Il carbocatione formato attacca quindi
il benzene, formando alla fine l’alchilbenzene. (L’alcole dopo la protonazione
perde acqua, trasformandosi poi in carbocatione).
5)
Formazione di acidi carbossilici aromatici
Gli
alchilbenzeni possono dare gli acidi carbossilici aromatici quando vengono
trattati con permanganato di potassio in ambiente acido e sotto riscaldamento:
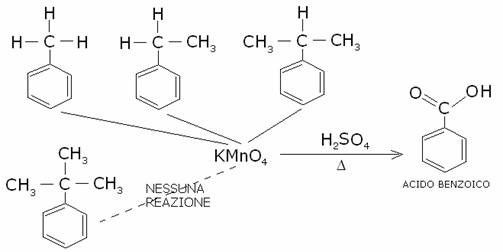
Come si vede,
gli alchilbenzeni col gruppo alchilico terziario legato all’anello resistono
all’ossidazione.
6) Acilazione
Avviene con
lo stesso meccanismo dell’alchilazione e dell’alogenazione:
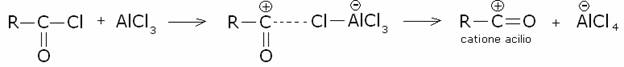
Il catione acilio è stabilizzato per risonanza:
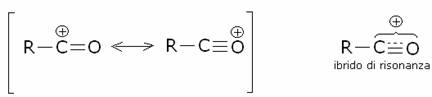
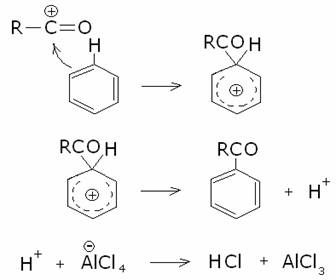
Il carbocatione acilico, più instabile di quello
alchilico, non presenta fenomeni di trasposizione.
7)
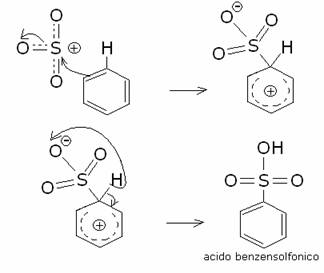
Solfonazione
La
sofonazione avviene per riscaldamento del benzene con acido solforico fumante,
contenente anidride solforica, che è l’elettrofilo che attacca il benzene. La
struttura dell’anidride solforica è:
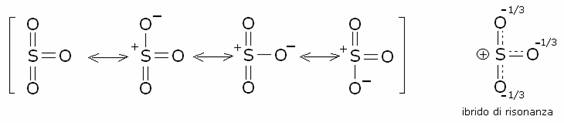
Il meccanismo della solfonazione è il seguente:
EFFETTO DEI SOSTITUENTI
Un gruppo legato al benzene può condizionare la
reattività dell’anello aromatico nei confronti di una successiva reazione di
sostituzione elettrofila. La reattività
è collegata alla velocità di reazione. I gruppi legati al benzene si
distinguono in:
gruppi attivanti,
che aumentano la velocità di
reazione, e quindi la reattività, e
in
gruppi disattivanti
che riducono la velocità di
reazione, e quindi la reattività.
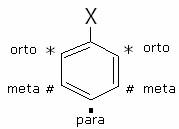
Oltre
all’effetto sulla velocità di reazione vi è anche l’effetto sulle posizioni
verso le quali il gruppo presente sull’anello benzenico orienta il gruppo entrante:
i gruppi
attivanti orientano i gruppi entranti (elettrofili) nelle posizioni orto e para;
i gruppi
disattivanti orientano i gruppi entranti (elettrofili) nelle posizioni meta.
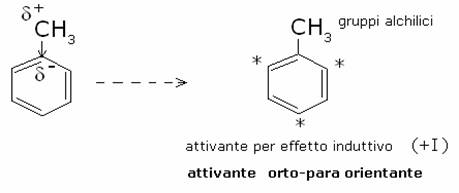
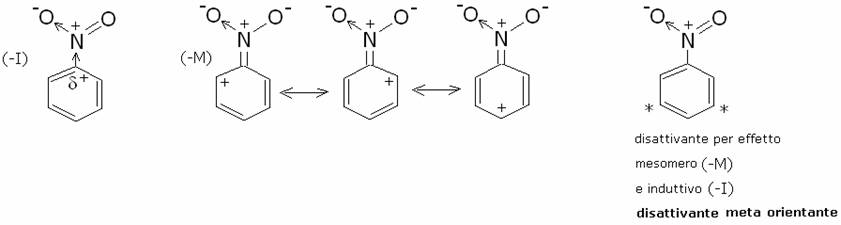
La natura attivante o disattivante di un gruppo viene
determinata in base al bilancio tra effetti induttivi (+ I o - I) e mesomeri (o
coniugativi) (+M o –M).
Esempi:
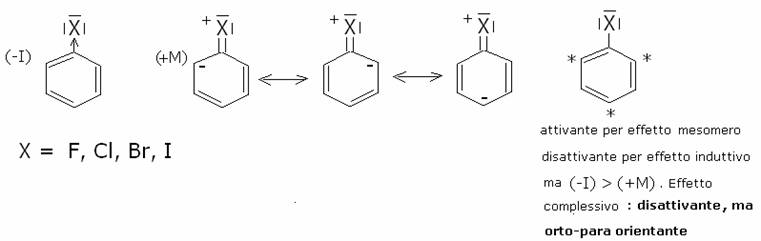
Nella tabella sottostante vengono elencate le
caratteristiche dei sostituenti più importanti:
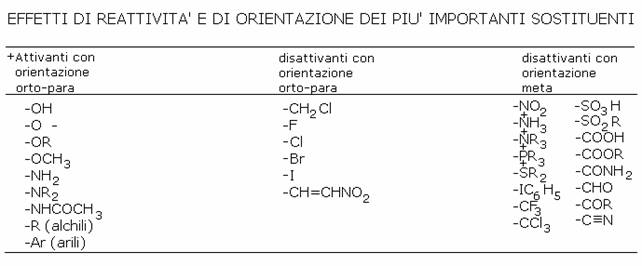
Vedi NOTA (1) a
pag. 13
GLI ALOGENURI ARILICI
1) Preparazione.
Abbiamo già
visto la reazione di alogenazione di Friedel-Kraft del benzene. Molto
importante per l’alogenazione è anche la reazione di Sandmeyer:
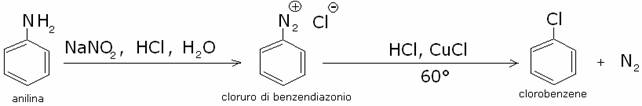
Il meccanismo procede attraverso radicali liberi e il
rame agisce come agente riducente e poi come
ossidante nelle fasi della reazione:
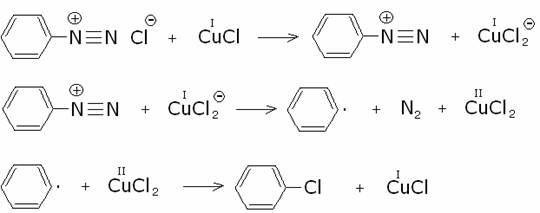
Il vantaggio della reazione di Sandmeyer è che
l’alogeno entra nell’anello benzenico nella stessa posizione prima occupata
dall’azoto, mentre nella reazione di Friedel-Kraft si possono formare miscele
di isomeri.
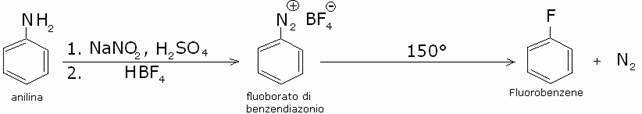
Nella florurazione, al posto del cloruro rameoso si
utilizza l’acido fluoborico:
2) Reazioni.
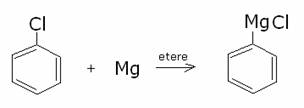
a) Formazione
dei reattivi di Grignard
Formano i
reattivi di Grignard, analogamente agli alogenuri alchilici.
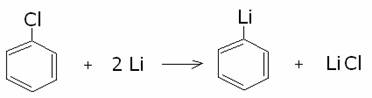
b) Reagiscono
col litio metallico per dare il fenil
litio
c) Reazioni di sostituzione nucleofila
aromatica di alogenuri aromatici attivati.
Il termine
“attivato” si riferisce alla presenza di uno o più gruppi elettronattrattori
legati all’anello, la cui funzione è quella di rendere più stabile l’intermedio
che si forma dopo l’attacco del nucleofilo, grazie ad una maggiore
delocalizzazione della carica negativa per risonanza.
Nell’esempio qui sotto il nitrogruppo permette appunto
una maggiore delocalizzazione della carica negativa e quindi una maggiore
stabilità dell’intermedio:
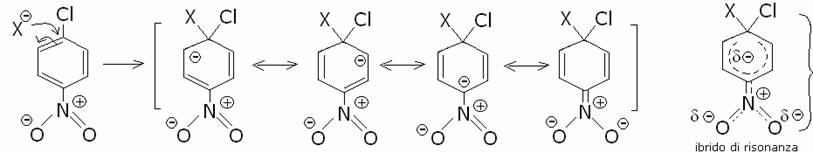
Successivamente lo ione Cl-
lascia l’anello, ricostituendo il sestetto aromatico del benzene
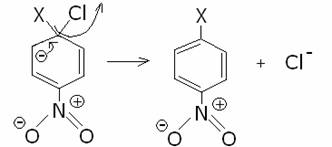
Altri
gruppi, come ad esempio -NO (nitroso), -CN
(cianuro), Diazonio (N2+), favoriscono la
sostituzione nucleofila aromatica.
d) Sostituzione
nucleofila aromatica mediante meccanismo di eliminazione-addizione.
Trattando un alogenuro alchilico con
una base molto forte si produce una
reazione di sostituzione con un meccanismo di eliminazione-addizione che passa
per un intermedio instabile: il benzino.
La
reazione globale è:

Il
meccanismo è il seguente:
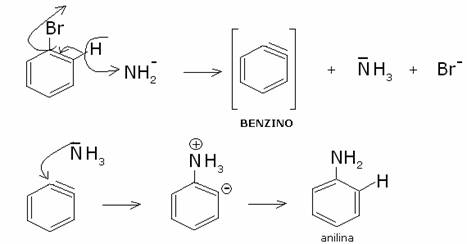
I SALI DI DIAZONIO E LORO
PRINCIPALI REAZIONI:
- REAZIONI DI
SOSTITUZIONE
- REAZIONE DI
COPULAZIONE
L’anilina
è una ammina primaria aromatica. Come tutte le ammine primarie, reagisce con
l’acido nitroso dando una N-nitrosoammina, che, in ambiente acido, forma un
sale di diazonio. Nel caso delle ammine primarie alifatiche tale composto ,
poco stabile, si dissocia in azoto elementare e in carbocatione che può legarsi
ad altri gruppi, formando diversi tipi di composti. Invece, nel caso delle
ammine aromatiche, il sale di diazonio è sufficientemente stabile e dà luogo a
diversi tipi di reazioni.
Formazione dei Sali di diazonio:
L’ammina
aromatica primaria (ad es. l’anilina) viene fatta reagire con acido nitroso in
ambiente acido. Si forma il catione nitrosonio, che avvia la reazione, secondo
i seguenti stadi:
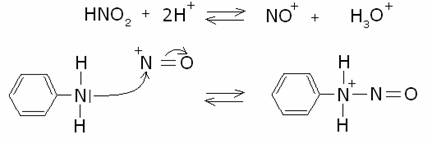
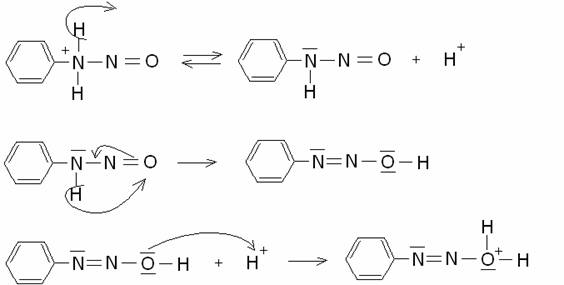
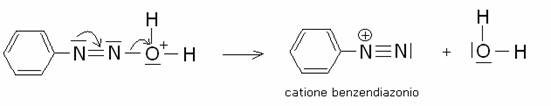
Il
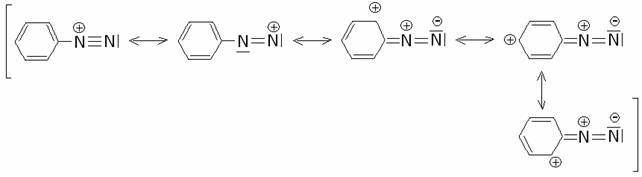
catione benzendiazonio è stabilizzato per risonanza:
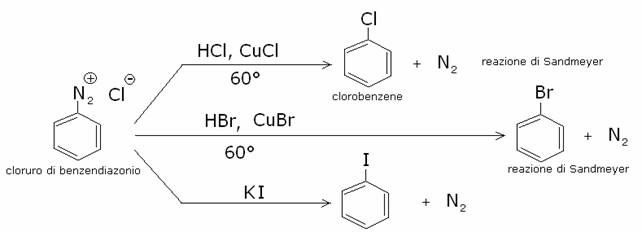
1) Reazioni di
sostituzione:
I Sali di
diazonio partecipano a numerose reazioni di sostituzione nelle quali il gruppo
diazonio, allontanandosi come azoto elementare, è facilmente sostituito da
diversi gruppi che si legano quindi all’anello:
a) Sostituzione
con alogeni (reazione di Sandmayer)
B) Reazione
di Schiemann:
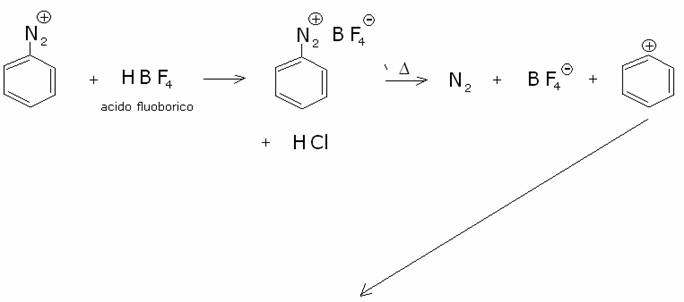
Formazione di fluorobenzene e di
composti complessi di varia natura
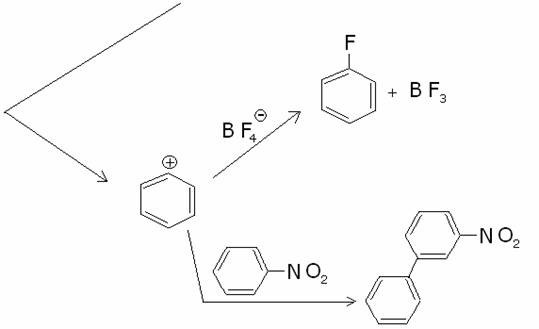
c) Formazione di cianobenzene (reazione di
Sandmeyer modificata):
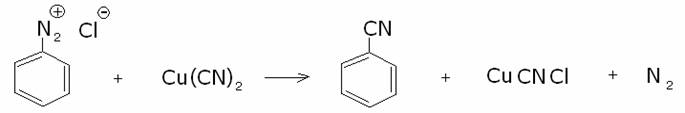
d)
Formazione di fenolo:
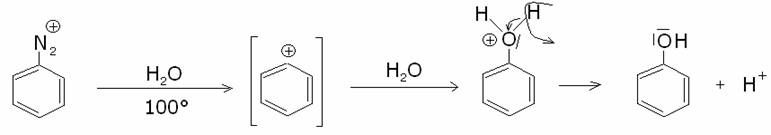
e) formazione di nitrobenzene (nitrazione):
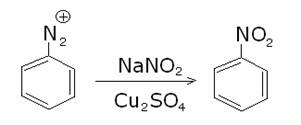
f) idrogenazione:
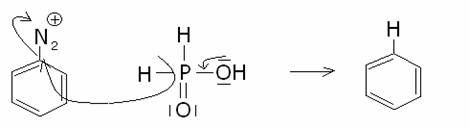
2)
Reazioni di Diazocopulazione:
E’ una
importantissima classe di reazioni di sostituzione elettrofila aromatica nelle
quali lo ione diazonio funge da elettrofilo (debole), attaccando un anello
aromatico attivato da gruppi fortemente elettrondatori (-OH, -NH2,
-NR2), producendo così composti fortemente colorati, importanti in
vari campi dell’industria. Consideriamo i seguenti esempi con una
arilammina e col fenolo (ione fenato):
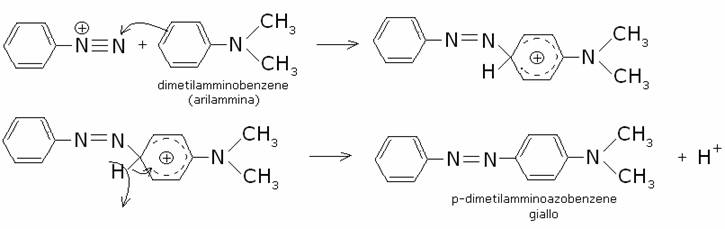
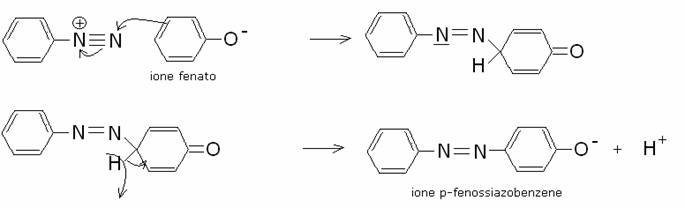
E’ da notare che la copulazione tra lo ione diazonio e
le arilammine deve avvenire in soluzioni il cui il pH è uguale o maggiore di 5.
Infatti per valori più piccoli di 5, le ammine si trasformano in ioni ammonio,
perdendo la caratteristica di forti attivanti dell’anello benzenico, non più
avendo l’azoto il suo doppietto elettronico disponibile per l’anello aromatico.
Il pH altresì non deve superare il valore di 9, poiché a valori molto alcalini
si verifica la seguente reazione che interessa lo ione benzendiazonio:
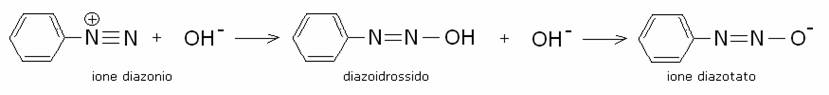
Né il diazoidrossido, né lo ione di azotato mostrano
carattere elettrofila, per cui non sono più capaci di attaccare un anello
aromatico, per quanto forte sia la sua attivazione. La necessità di mantenere
il pH al disotto o uguale a 9 è fondamentale quando la copulazione avviene col
fenolo.
Quest’ultimo è un acido debole (Ka = 10-10
):
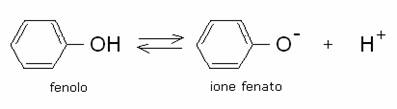
Il gruppo OH del fenolo è un attivante di media forza
quando è associato. Diventa un forte attivante, e quindi può copulare con lo
ione diazonio, quando è dissociato (ione fenato). La dissociazione però diventa
significativa solo a valori di pH uguali o superiori a 8. La copulazione col
fenolo perciò deve avvenire a pH compreso tra 7 e 8. Al di là di 9 avvengono le
trasformazioni su descritte dello ione diazonio che ne annullano il carattere
elettrofilo, impedendo la copulazione. La velocità della reazione di
diazocopulazione è perciò molto sensibile al pH dell’ambiente in cui avviene la
reazione.
NOTA(1) di pag 6:
Riguardo agli alchilbenzeni, è importante sottolineare
che il metilbenzene presenta una reattività superiore agli altri gruppi
alchilici. Mettiamo, ad esempio, a confronto la reattività del metilbenzene con
quella del terzbutilbenzene. In quest’ultimo, essendoci il radicale terzbutile,
dovremmo aspettarci una maggiore attivazione dell’anello benzenico, poiché
l’effetto induttivo del terzbutile è maggiore di quello relativo al metile,
essendo il terzbutile un radicale terziario:
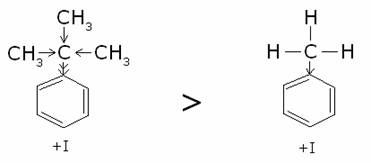
In realtà risulta più attivato il metilbenzene, che,
in particolare, orienta l’elettrofilo entrante preferibilmente nella posizione
para. Questa apparente anomalia viene spiegata col fenomeno dell’iperconiugazione che coinvolge i legami
C-H del metile. Nella figura sottostante, oltre all’ibrido di risonanza che si
riferisce alle tre formule limiti con la carica positiva situata nelle
posizioni orto e para, vengono riportate anche le tre formule limiti relative
alla iperconigazione che coinvolge
il gruppo metile.
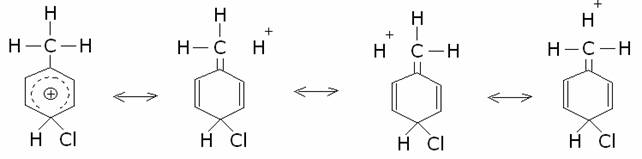
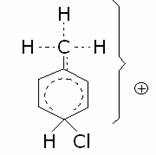
Come si nota dall’ibrido di risonanza riportato qui
sotto, la più estesa delocalizzazione
della carica positiva produce una più elevata stabilità dell’intermedio
carbocationico, e quindi una maggiore reattività del metilbenzene:
.
I FENOLI
1) Sintesi e
proprietà fisiche dei fenoli.
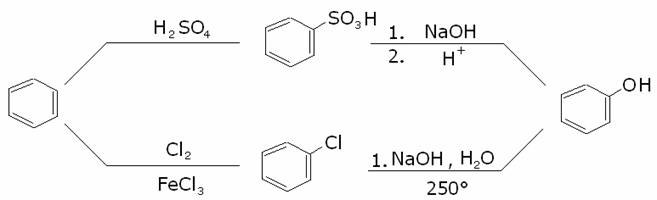
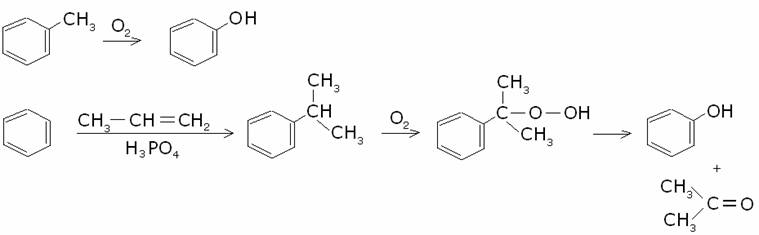
Per quest’ultima reazione, il relativo meccanismo è
riportato tra le reazioni di sintesi dei composti carbonilici..
2) Reazioni che
coinvolgono il legame O-H del fenolo.
Il fenolo ha
una energia di stabilizzazione (energia di risonanza) di 40Kcal/mole, 38 delle quali dovute
all’anello benzenico. Le 2 Kcal/mole rimanenti sono dovute alla delocalizzazione
di uno dei due doppietti esterni dell’ossigeno che viene condiviso con l’anello
benzenico:
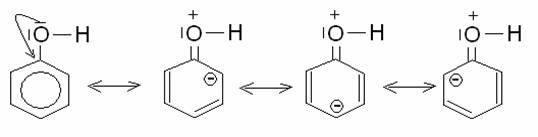
La conversione del fenolo in ione fenato porta
anch’essa ad una notevole energia di stbilizzazione, addirittura un po’ più
elevata del fenolo stesso.
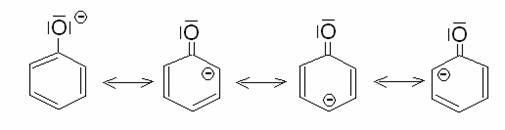
Come si vede dalle formule limiti di risonanza, ciò è
dovuto al fatto che in esse non vi è separazione di carica, che invece è
presente in quelle relative al fenolo indissociato. Il fenolo quindi è un acido
molto più forte degli alcoli (Ka fenolo =10 -10, Ka alcoli
= 10 -16).
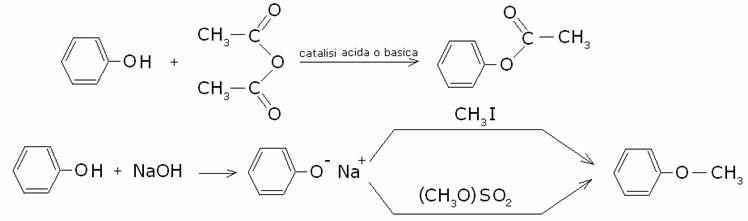
L’acidità del fenolo gli consente di preparare ad
esempio esteri per reazione con gli acidi carbossilici, eteri per reazione con
alogenuri alchilici o con esteri dell’acido solforico:
3) Reazioni
sull’anello aromatico del fenolo.
Il fenolo, e
ancor più lo ione fenato, a causa dell’eccesso di carica negativa che risiede
nell’anello è un forte attivante nelle reazioni di sostituzione elettrofila
all’anello. Infatti, ad esempio, nella reazione di bromurazione, alla fine
vengono attaccate tutte le posizioni orto e para:
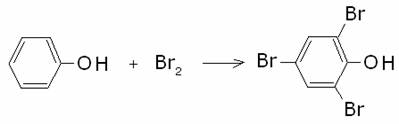
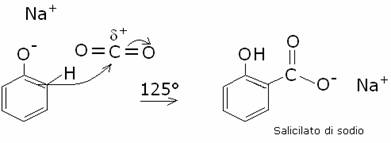
La forte reattività dello ione fenato si manifesta
nella possibilità dell’attacco all’anello benzenico di elettrofili anche molto
deboli come l’anidride carbonica. E’ questa la reazione di Kolbe:
Nella reazione
di Reimer-Tiemann è il cloroformio ad attaccare l’anello dello ione fenato,
dando alla fine l’aldeide salicilica:
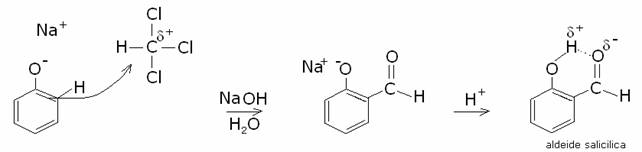
Viene favorito l’attacco in para, poiché in tal modo
si forma un legame ad idrogeno intramolecolare, che stabilizza l’intera
molecola.
4) Ossidazione
dei fenoli.
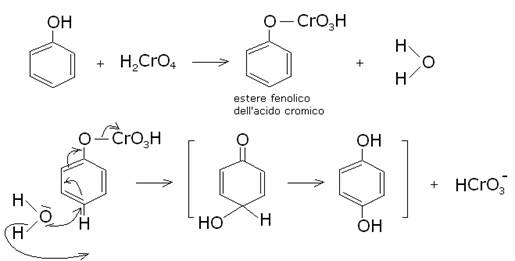
L’ossidazione
del fenolo per acido cromico porta al para-benzochinone:
POLIFENOLI
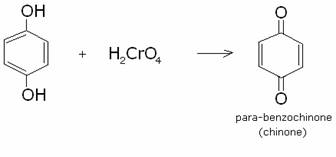
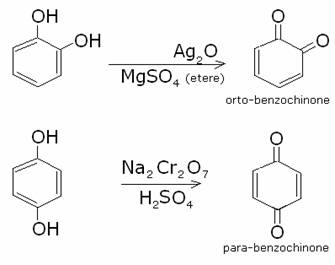
Alcuni polifenoli importanti:

I polifenoli con due gruppi ossidrilici in orto o in
para subiscono facilmente ossidazioni a chinoni:
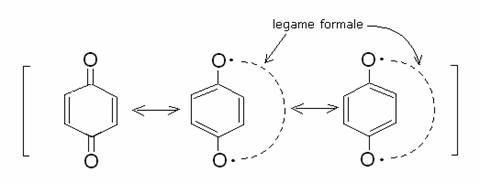
Strettamente parlando, i chinoni sono dei di chetoni
ciclici, più che composti armoatici. Sono abbastanza stabili, grazie alla
stbilizzazione per risonanza (di circa 5 Kcl/mole) dovuta alle seguenti formule
limiti di risonanza (per il para-benzo chinone):
Reazioni redox dei chinoni.
La caratteristica più importante dei chinoni è la
riduzione ai corrispondenti composti diidrossiaromatici:
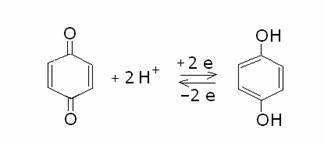
Tali riduzioni sono inusuali tra i composti organici,
essendo sufficientemente rapide, reversibili e riproducibili, dando così luogo
a potenziali elettrodici facilmente riproducibili in una cella elettrolitica.
Il potenziale della coppia chinone – idrochinone è sensibile al pH della
soluzione e varia di 0,059 volt per la variazione di una unità pH. Mescolando
soluzioni alcoliche equimolari di idrochinone e di chinone si forma un
precipitato verde scuro costituito da un complesso 1:1 dei due composti. Si
tratta di un complesso a trasferimento di carica, nel quale il donatore (di
carica negativa) è l’idrochinone, l’accettore è il chinone.
IDROCARBURI AROMATICI POLINUCLEARI
I Più importanti
sono:
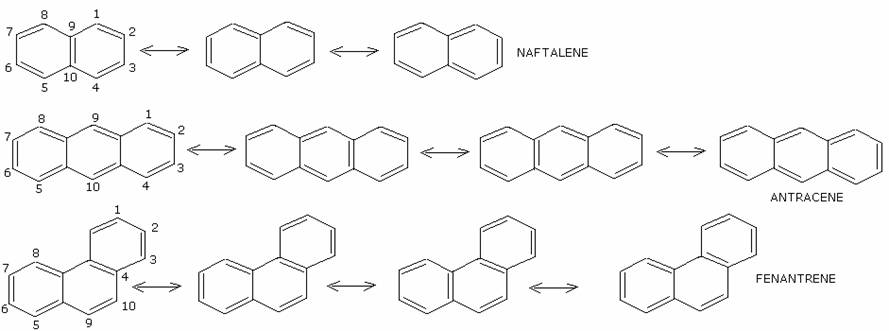
Sono più reattivi del benzene rispetto alle reazioni
di sostituzione elettrofila e di addizione, poiché le energie di
stabilizzazione per risonanza sono inferiori a quella del benzene nell’ordine
neaftalene >fenantrene>antracene. Dalle formule di risonanza si osserva
infatti che non tutti i legami sono uguali rispetto all’ordine di legame.
Infatti, mentre nel benzene ogni legame ha ordine 1,5, e la stessa lunghezza,
nel naftaline, antracene e fenantrene le lunghezze dei legami non sono tutte
uguali. Ad es. nel nafatalene la lunghezza del legame 1-2 ( 1,36 Ǻ) è più
piccola di quella dei legami 2-3 e 9-10 ( 1,42 Ǻ). Infatti, come si vede
dalle formule di risonanza il legame 1-2 è sede per due volte su tre di un
doppio legame, a fronte di una volta su tre per il legame 2-3.
1) Reazioni di
sostituzione.
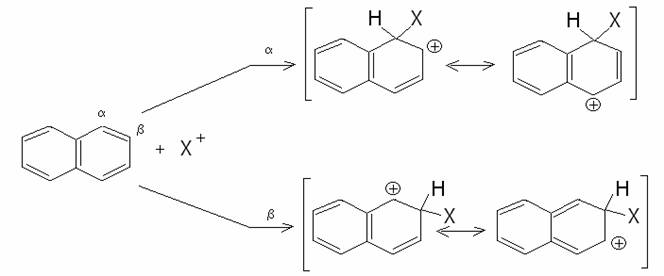
a) Naftalene.
Nelle reazioni
di sostituzione elettrofila aromatica del naftalene le posizioni a sono molto più reattive delle posizioni b:
Il motivo risiede nel fatto che l’attacco dell’elettrofilo
in una posizione conduce a formule di
risonanza che vedono la carica elettrica delocalizzata sull’anello ove si è
verificato l’attacco elettrofila, senza intaccare l’aromaticità dell’altro
anello, mentre un attacco in una posizione
intacca l’aromaticità dell’altro anello:
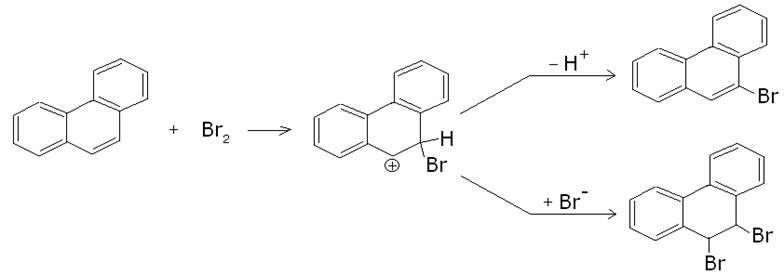
b) Fenantrene.
Nel
fenantrene, più reattivo del naftalene, il bromo elementare reagisce
direttamente, attaccando la posizione
9 o 10, che fornisce l’intermedio carbocationico più stabile, portando ad una
sostituzione o ad una addizione:
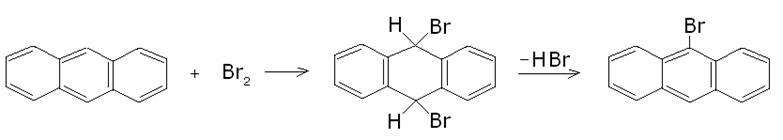
c) Antracene.
L’antracene,
ancora più reattivo del fenantrene ha una tendenza ad addizionare nelle
posizioni 9 e 10, più che partecipare a reazioni di sostituzione. Comunque i
prodotti di addizione della nitrazione e della alogenazione si trasformano
rapidamente, dando complessivamente un prodotto di sostituzione nella posizione
9:
2) Reazioni di addizione (riduzione).
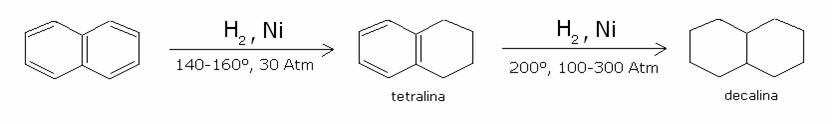
a) Naftalene.
Il naftalene può essere ridotto a tetraedronaftalene o
tetralina con idrogeno in presenza di Nichel. Per una più prolungata
esposizione all’idrogeno può essere interamente ridotto a peridronaftalene o
decalina.
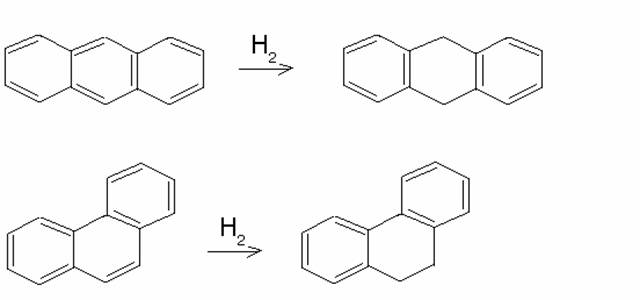
b) Antracene e
Fenantrene.
Sono ridotte molto facilmente a diidroderivati con
idrogeno molecolare, nelle posizioni 9 e 10,
rispettivamente a 9,10-diidroantracene e a 9,10 diidrofenantrene
2) Reazioni di
ossidazione.
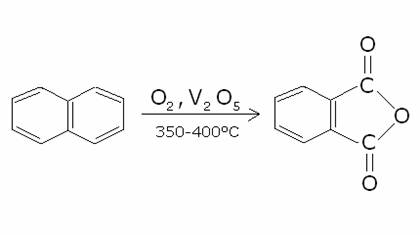
a)
Naftalene.
E’ ossidato ad
alta temperatura dall’ossigeno molecolare in presenza di pentossido di di
vanadio, dando come prodotto di reazione l’anidride ftalica:
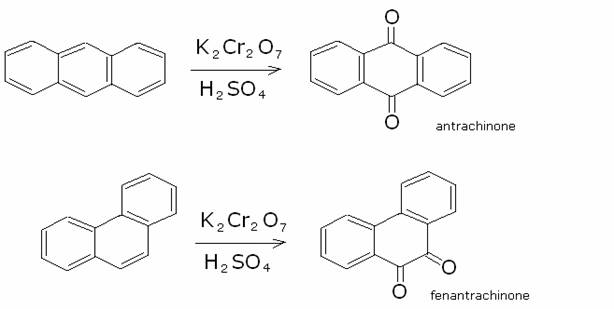
b) Antracene e
Fenantrene.
Sono più facilmente ossidabili del naftalene. Infatti
reagiscono a temperatura ambiente con comuni ossidanti, come bicromato di
potassio e acido solforico:
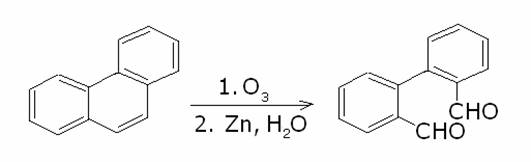
Il fenantrene, trattato con ozono, quindi con zinco in
acqua, forma la 2, 2’-difenilaldeide:
Torna a ARGOMENTI
DI CHIMICA ORGANICA
